56. Nucleotide Degradation: Biochemical Pathways and Clinical Pathologies
Welcome to this comprehensive overview of cellular metabolism and biochemical pathology! The core purpose of this complete slide deck is to thoroughly demystify the biochemical pathways and clinical pathologies associated with nucleotide breakdown. From fundamental enzymatic reactions to devastating genetic disorders, this visual guide will illuminate the profound impact of molecular recycling and waste management on human health.
Slide 1: Nucleotide Degradation: The Crucial Link Between Biochemistry and Human Pathology
A beautiful crystal can be a marvel of nature, but inside a human joint, it becomes an agent of agonizing pain. The core purpose of this slide is to introduce the fascinating intersection of molecular biology and human disease.
When studying the overarching concept of nucleotide degradation, one must understand that it is never just about waste disposal. It represents a highly regulated biochemical framework that carefully balances the synthesis and breakdown of essential cellular components. The process of nucleotide degradation ensures that excess purines and pyrimidines are safely processed. However, when these pathways fail, the results are clinically profound.
The title slide visually anchors this topic with a striking representation of a purine ring transforming into a sharp, jagged crystal. This imagery directly previews the most common clinical consequence of improper nucleotide degradation: the crystallization of uric acid. By examining the structural breakdown of these vital molecules, medical and college students can begin to appreciate the delicate metabolic balance required for human health.
Understanding nucleotide degradation provides a foundational map for the entire slide deck. It prepares the learner to transition from basic molecular structures to complex enzymatic cascades. Ultimately, mastering the principles of nucleotide degradation empowers future healthcare professionals to accurately connect subtle genetic mutations with severe, systemic clinical disorders.
Slide 2: Nucleotide Degradation and Digestion: How Cells Manage Energetically Expensive Investments
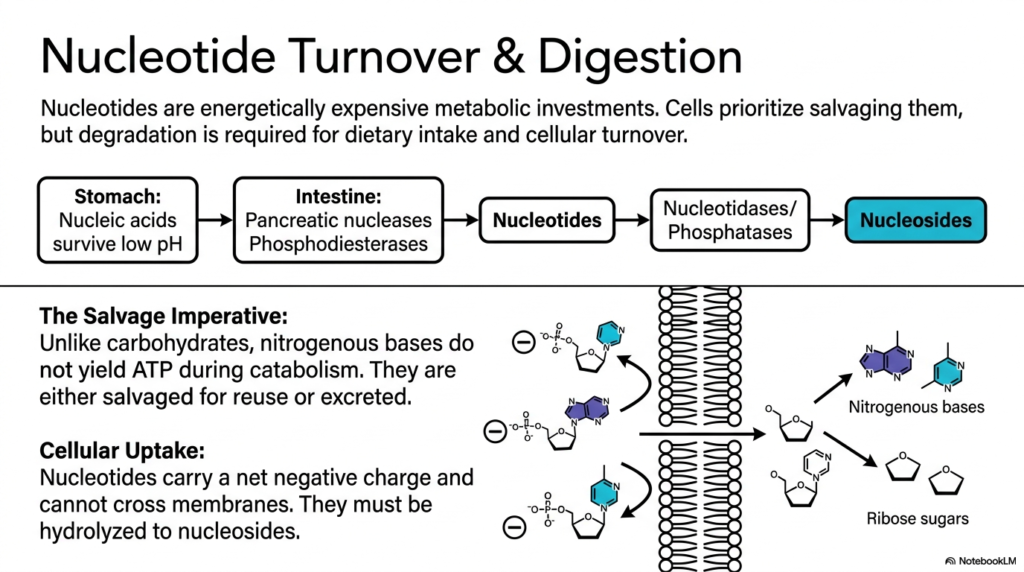
Imagine throwing away a perfectly good diamond just because it has a slight scratch. Cells face a similar dilemma with their genetic material. This slide explains how the body digests dietary nucleic acids and highlights the cellular imperative to salvage rather than waste them.
The process of nucleotide degradation begins long before individual cells manage their internal waste. It begins in the digestive tract, where dietary nucleic acids withstand the stomach’s harsh, acidic environment. In the intestine, pancreatic nucleases and phosphodiesterases break these macromolecules down into individual nucleotides. However, these molecules carry a net negative charge, making cellular uptake impossible without further modification.
To cross the lipid bilayer, nucleotides must be hydrolyzed into neutral nucleosides by specific nucleotidases and phosphatases. This necessary step highlights a crucial rule of nucleotide degradation: cellular transport requires the removal of bulky, charged phosphate groups. Once inside the cell, these nitrogenous bases face a critical metabolic crossroads. They will either be recycled or committed to complete breakdown.
Unlike the catabolism of carbohydrates or lipids, the catabolism of nitrogenous bases does not yield ATP. Because creating them from scratch takes massive amounts of energy, cells prioritize salvage pathways. Therefore, cellular nucleotide degradation is generally a pathway of last resort, utilized only when dietary intake or cellular turnover strongly exceeds the immediate need for building blocks.
Slide 3: Nucleotide Degradation Context: The Biosynthetic Economy of De Novo vs. Salvage Pathways
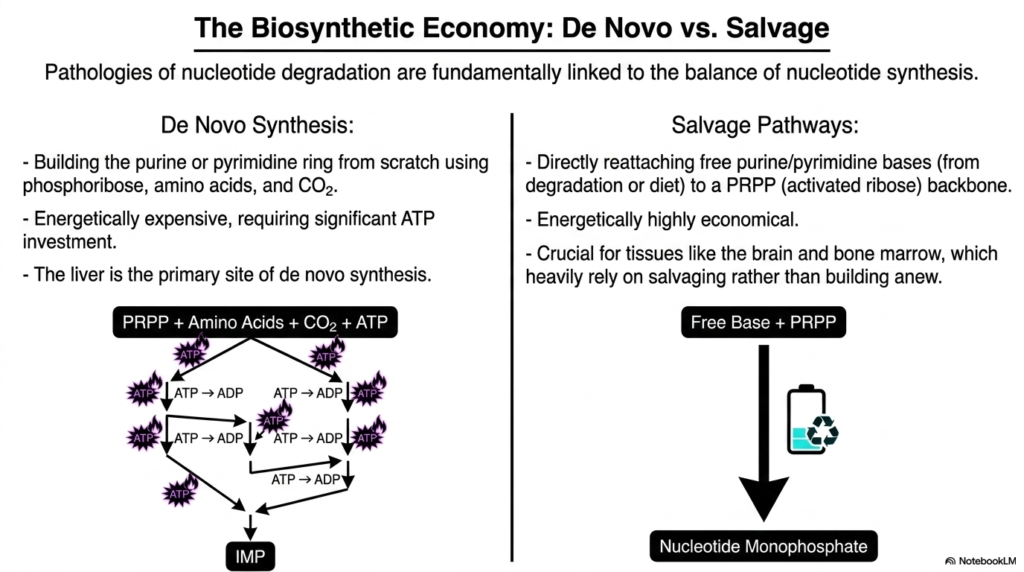
Building a house from raw timber is exhausting, but renovating an existing frame is highly efficient. Cells employ the exact same logic for their genetic material. This slide explores the immense energetic difference between synthesizing bases from scratch and recycling them.
To truly understand the consequences of nucleotide degradation, one must first grasp how these complex molecules are built. De novo synthesis is an energetically demanding process that constructs purine and pyrimidine rings completely from scratch. Using phosphoribose, amino acids, and carbon dioxide, the liver invests heavily in ATP to generate intermediate molecules like IMP.
Conversely, salvage pathways offer a highly economical alternative. By directly reattaching free purine or pyrimidine bases to an activated ribose backbone, the cell bypasses the massive ATP expenditure of de novo synthesis. Pathologies associated with nucleotide degradation are fundamentally linked to the delicate balance between these two synthetic routes.
Certain tissues, such as the brain and bone marrow, lack robust de novo machinery and rely almost exclusively on salvaging free bases. When genetic defects force these tissues to rely entirely on nucleotide degradation instead of recycling, devastating clinical symptoms quickly emerge. Recognizing this biosynthetic economy is essential for predicting how disruptions in nucleotide degradation impact different organ systems.
Slide 4: Purine Nucleotide Degradation: The Biochemical Convergence on Xanthine
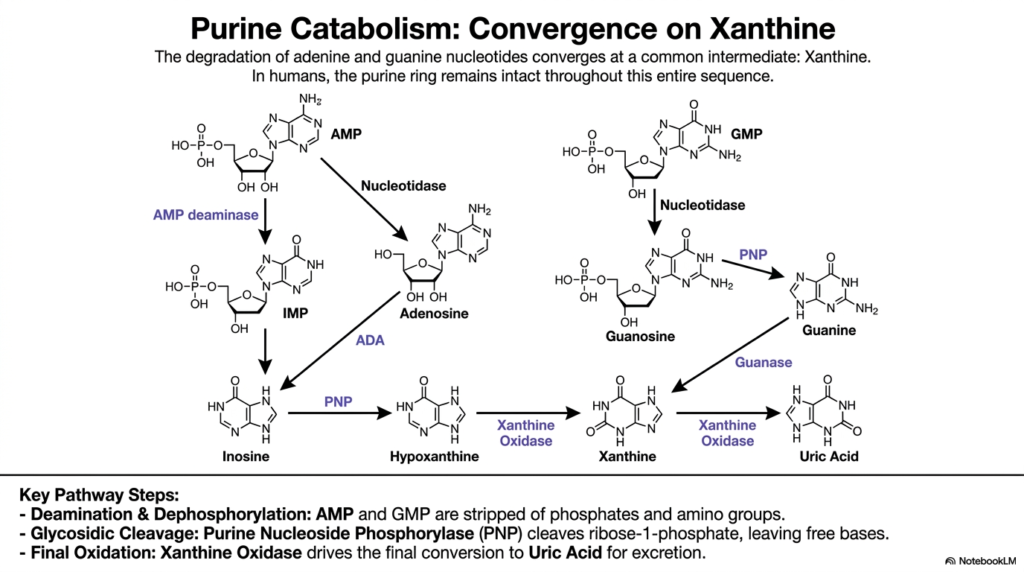
All roads lead to Rome, and in human purine catabolism, all molecular pathways converge on a single intermediate. The core purpose of this slide is to trace the step-by-step enzymatic breakdown of adenine and guanine.
The complex cascade of purine nucleotide degradation is an elegant exercise in molecular dismantling. Interestingly, in humans, the purine ring structure remains entirely intact throughout this precise sequence. The pathway naturally begins with dephosphorylation, where AMP and GMP are stripped of their bulky phosphate groups by nucleotidases to form nucleosides.
Next, AMP undergoes deamination via AMP deaminase to form IMP, or adenosine is deaminated by Adenosine Deaminase to form inosine. A critical enzyme, Purine Nucleoside Phosphorylase, then steps in. During this specific phase of nucleotide degradation, this enzyme cleaves ribose-1-phosphate, thereby safely liberating the free nitrogenous bases hypoxanthine and guanine.
Both of these bases are then swiftly funneled toward a common molecular fate. Guanase converts guanine to xanthine, while hypoxanthine is chemically oxidized to xanthine. This funneling effect vastly simplifies purine nucleotide degradation, ultimately relying on the enzyme Xanthine Oxidase to drive the final oxidation step, converting xanthine into sparingly soluble uric acid for eventual excretion.
Slide 5: The Engine of Nucleotide Degradation: Xanthine Oxidase and its Molybdopterin Complex
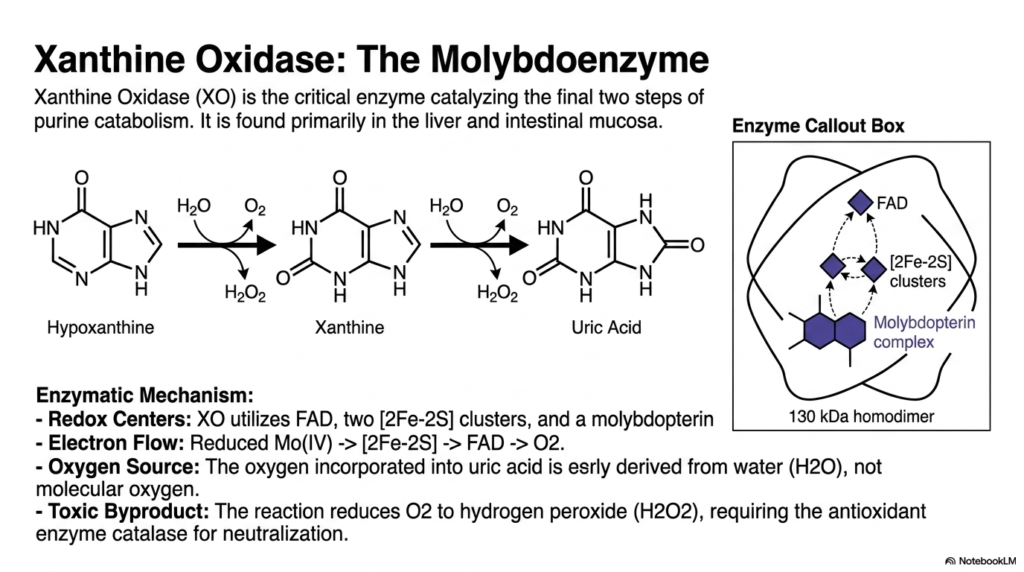
Some cellular machines require exotic parts to function correctly. Xanthine Oxidase relies heavily on the trace metal molybdenum to carry out its final, crucial reactions. This slide breaks down the intricate redox mechanisms of this vital enzyme.
Xanthine Oxidase is the undisputed powerhouse driving the final stages of purine nucleotide degradation. Located primarily in the liver and intestinal mucosa, this massive 130-kDa homodimeric enzyme catalyzes the conversion of hypoxanthine to xanthine and of xanthine to uric acid. Its complex enzymatic mechanism relies on multiple functional redox centers to shuttle electrons efficiently.
The enzyme functionally utilizes Flavin Adenine Dinucleotide, two iron-sulfur clusters, and a highly specialized molybdopterin complex. During nucleotide degradation, electron flow moves smoothly from reduced molybdenum to the iron-sulfur clusters, then to the flavin center, and finally to molecular oxygen. Interestingly, the oxygen incorporated into the uric acid molecule is actually derived from surrounding water, not dissolved oxygen gas.
This powerful step in nucleotide degradation comes with a very dangerous trade-off. The chemical reaction reduces molecular oxygen to hydrogen peroxide, a highly reactive and toxic oxygen species. Therefore, this specific phase of purine nucleotide degradation must always be tightly coupled with the antioxidant enzyme catalase, which neutralizes the dangerous hydrogen peroxide byproduct into harmless water and oxygen.
Slide 6: The Evolutionary Quirks of Nucleotide Degradation: The Fate of Uric Acid
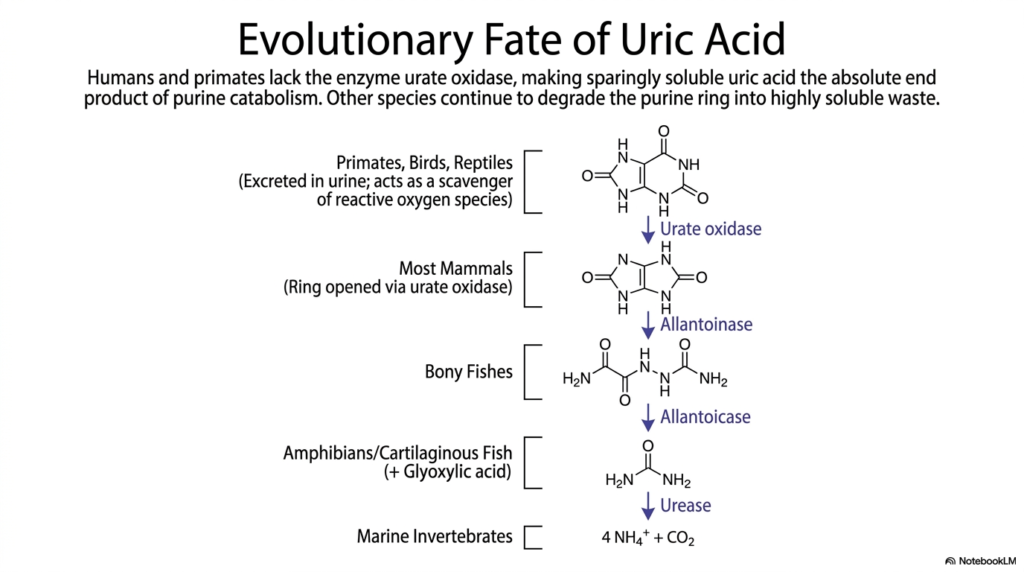
Why do humans commonly suffer from gout while dogs and fish do not? Evolution simply left primates with a missing enzymatic link. This slide explores the fascinating comparative biochemistry of urate processing across different biological species.
The absolute endpoint of purine nucleotide degradation varies wildly across the animal kingdom. While many organisms gracefully continue to dismantle the purine ring into highly soluble waste products, humans and higher primates experience a hard stop. We lack the functional enzyme urate oxidase, making uric acid the absolute end product of our catabolic pathway.
In most other mammals, urate oxidase successfully cleaves the purine ring to form allantoin, which is easily dissolved and excreted in urine. Bony fishes take nucleotide degradation even further, using enzymes such as allantoinase and allantoicase to produce simple urea. Marine invertebrates completely break the molecule down into basic ammonia and carbon dioxide.
Because humans inherently stop at uric acid, we constantly operate near the dangerous solubility limit of this molecule in our blood. However, this evolutionary halt in nucleotide degradation might offer a surprising survival advantage. Uric acid acts as a powerful scavenger of reactive oxygen species, potentially protecting primates from oxidative stress despite the severe risk of crystallization.
Slide 7: Skeletal Muscle and Nucleotide Degradation: Anaplerosis via the Purine Nucleotide Cycle
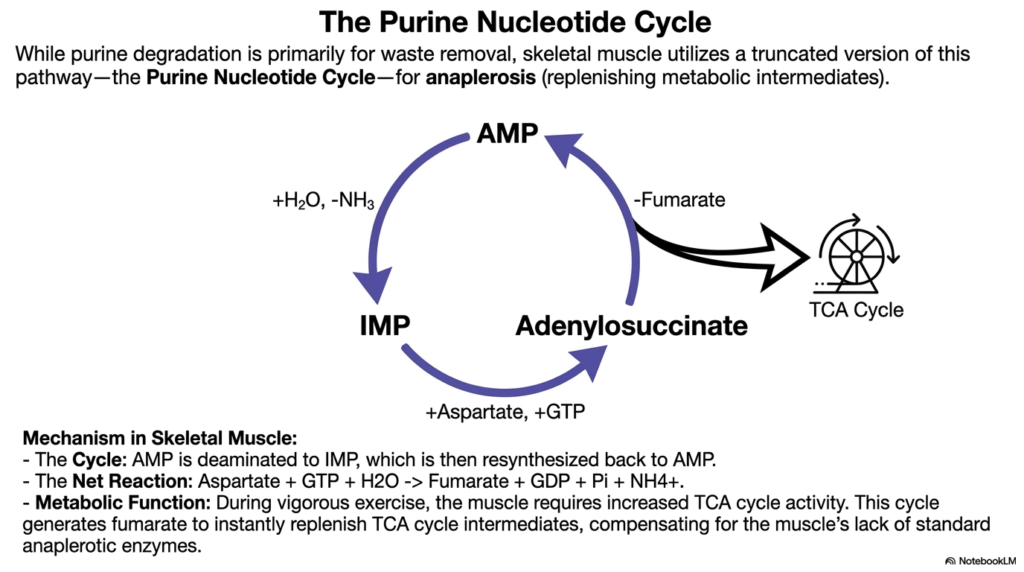
Skeletal muscle works incredibly hard and burns fuel fast, requiring instant metabolic replenishment. This slide brilliantly demonstrates how muscle tissue cleverly hijacks a standard waste pathway to keep cellular energy production spinning.
While the complete nucleotide degradation process is primarily designed for cellular waste removal, skeletal muscle utilizes a truncated, cyclical version of this pathway for an entirely different purpose. This localized loop, officially known as the Purine Nucleotide Cycle, performs crucial anaplerotic functions. Anaplerosis refers to the rapid metabolic replenishment of intermediates that keep central energy cycles safely running.
In this highly modified form of nucleotide degradation, AMP is safely deaminated to form IMP. Instead of continuing straight down the waste pathway, IMP is rapidly resynthesized back into AMP via an adenylosuccinate intermediate. The net chemical reaction requires aspartate and GTP but, crucially, releases fumarate as a valuable byproduct.
During vigorous physical exercise, the muscle rapidly depletes its intermediates and desperately requires increased TCA cycle activity. Because muscle tissue naturally lacks standard anaplerotic enzymes found in the liver, it relies on this specific segment of nucleotide degradation. Generating fumarate instantly restocks the TCA cycle, allowing the muscle to maintain high levels of ATP production under intense physical stress.
Slide 8: Pyrimidine Nucleotide Degradation: The Ring Cleavage Strategy
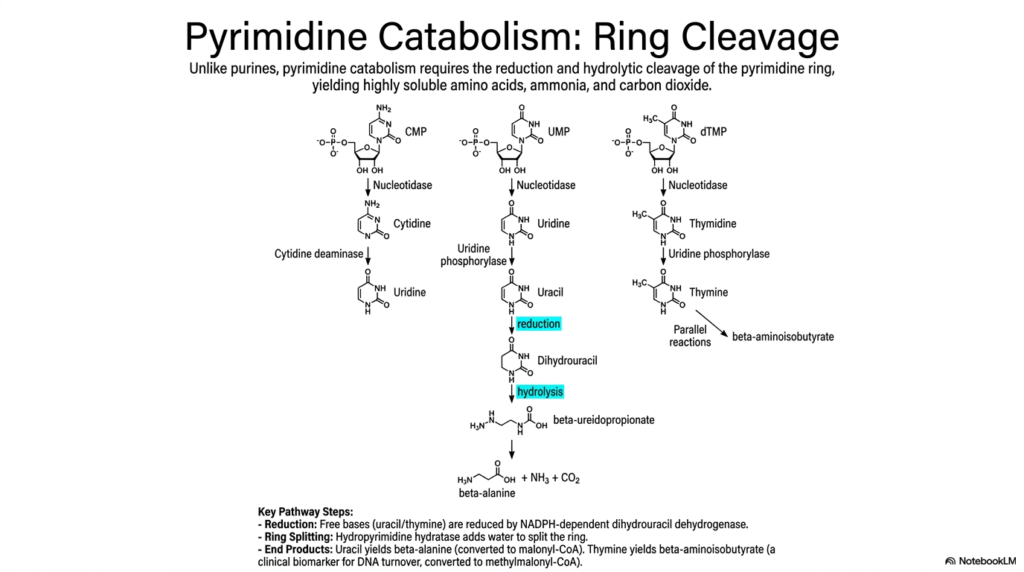
Unlike the incredibly stubborn purine ring, pyrimidine rings are easily cracked open and dismantled. This slide contrasts the structural fate of pyrimidines, highlighting a completely different biochemical strategy for managing cellular waste.
The pathway of pyrimidine nucleotide degradation stands in stark contrast to that of purine nucleotides. Rather than meticulously preserving the ring structure, pyrimidine catabolism requires complete chemical reduction and hydrolytic cleavage of the molecule. The process begins similarly, with nucleotidases stripping phosphate groups from CMP, UMP, and dTMP to form standard nucleosides.
Following gentle deamination and the removal of the ribose sugar, the free bases uracil and thymine enter the core phase of pyrimidine nucleotide degradation. An NADPH-dependent enzyme, dihydrouracil dehydrogenase, chemically reduces the free bases. Next, hydropyrimidine hydratase adds water to successfully split the ring wide open, a defining physical feature of this particular pathway.
The final products of pyrimidine nucleotide degradation are highly water-soluble, so they rarely cause clinical pathologies related to precipitation. Uracil ultimately yields beta-alanine, which smoothly converts to malonyl-CoA, while thymine yields beta-aminoisobutyrate. These highly soluble amino acids, along with ammonia and carbon dioxide, are readily cleared or safely repurposed by the body.
Slide 9: Comparing Purine and Pyrimidine Nucleotide Degradation Strategies
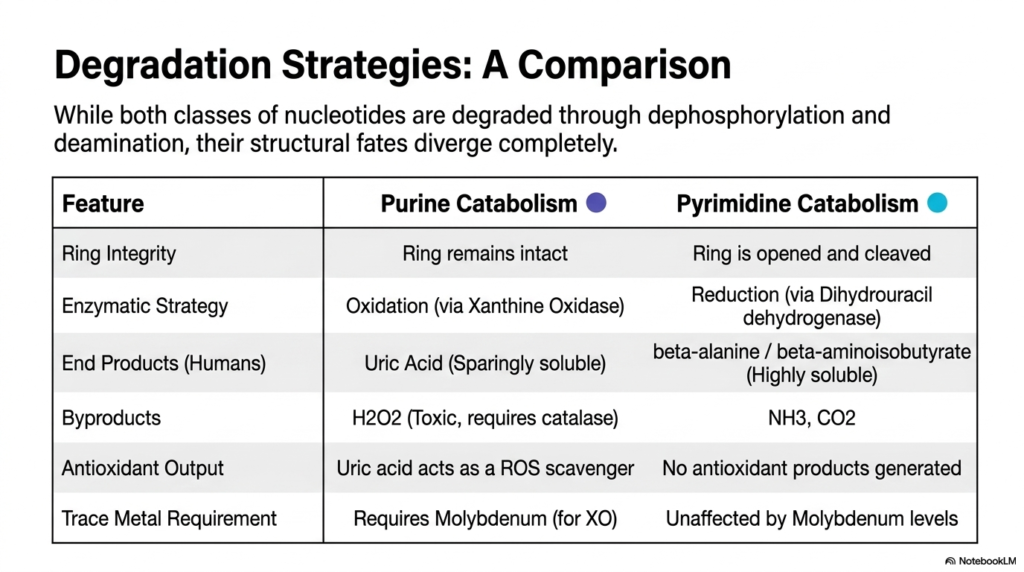
Two molecular families, yet two radically different cellular fates. This slide provides an excellent high-level comparative summary of how cells manage their various genetic building blocks.
A side-by-side analysis of nucleotide degradation strategies uncovers a fascinating evolutionary divergence. While both classes of molecules initially undergo similar dephosphorylation and deamination, their ultimate structural fates diverge completely. Purine catabolism focuses intensely on oxidation, diligently keeping the multi-ring structure entirely intact all the way to excretion.
In sharp contrast, pyrimidine nucleotide degradation successfully employs a bold strategy of chemical reduction and aggressive ring cleavage. This fundamental difference strictly dictates the physical properties of the resulting biological waste. Purine breakdown leaves humans with sparingly soluble uric acid, while pyrimidine breakdown yields highly soluble beta-amino acids that pose virtually no biological risk of crystallization.
Furthermore, the biochemical byproducts of these two pathways differ significantly. Purine nucleotide degradation unfortunately generates toxic hydrogen peroxide and strictly requires trace metals like molybdenum for normal Xanthine Oxidase function. However, it thoughtfully provides a systemic antioxidant in the form of uric acid. Pyrimidine degradation produces only ammonia and carbon dioxide, without generating any systemic antioxidants.
Slide 10: Pathologies of Nucleotide Degradation: The Clinical Reality of Hyperuricemia and Gout
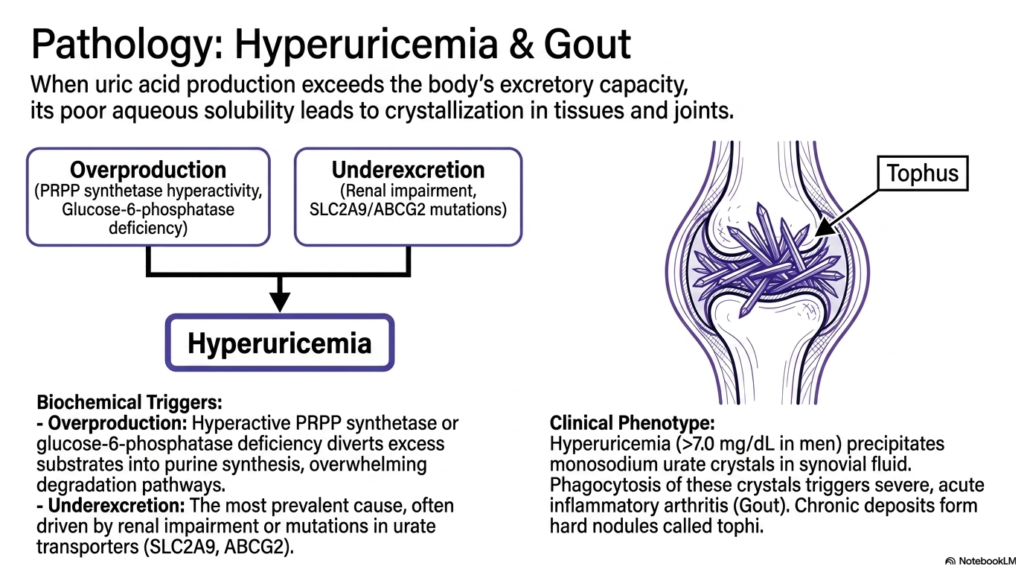
When biological waste management utterly fails, soft tissues rapidly turn into inflammatory battlegrounds. This slide visually illustrates how a dangerous imbalance in uric acid levels leads to the painful, needle-like crystals that characterize gout.
The clinical consequences of disrupted nucleotide degradation most commonly manifest as hyperuricemia, defined as serum uric acid levels exceeding 7.0 mg/dL in men. This severe condition arises when uric acid production vastly exceeds the body’s extremely limited excretory capacity. The fundamental root causes are generally divided into two main clinical categories: biochemical overproduction or renal underexcretion.
Overproduction frequently occurs when genetic mutations hyperactivate enzymes like PRPP synthetase or when glucose-6-phosphatase is deficient. These issues inappropriately divert large amounts of excess substrate into purine synthesis, thereby overwhelming the standard pathways of nucleotide degradation. However, the most prevalent cause of hyperuricemia remains underexcretion, often driven by failing kidneys or inherited mutations in specific urate transporters.
When this severe bottleneck in nucleotide degradation occurs, the sparingly soluble uric acid precipitates violently from the blood. It forms sharp monosodium urate crystals directly in joint synovial fluid. Phagocytic immune cells furiously attack these crystals, triggering the severe, acute inflammatory arthritis medically recognized as gout.
Slide 11: Pharmacological Modulation of Nucleotide Degradation: Allopurinol
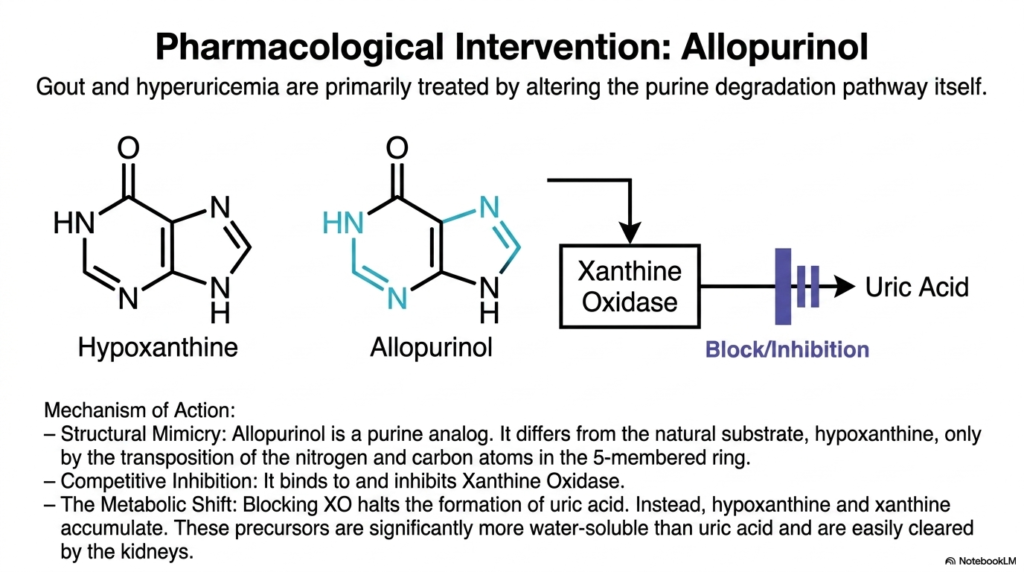
Sometimes the absolute best way to stop a runaway train is to chemically block the tracks. This slide elegantly introduces the brilliant pharmacological mimicry that safely halts uric acid production at its source.
Treating severe gout often requires direct, precise interference with the complex pathways of nucleotide degradation. The gold-standard medication commonly utilized for this is allopurinol. This incredibly powerful drug fundamentally exerts its effects through clever structural mimicry. Allopurinol is a synthetic purine analog that appears nearly identical to the natural substrate, hypoxanthine.
The only real structural difference is a simple transposition of the nitrogen and carbon atoms deeply within the five-membered ring. Because of this extreme similarity, allopurinol effectively acts as a highly competitive inhibitor during nucleotide degradation. It binds tightly and securely to the active site of Xanthine Oxidase, effectively blocking the vital enzyme from converting normal substrates into uric acid.
By specifically inhibiting nucleotide degradation at this highly specific enzymatic step, allopurinol induces a profound metabolic shift. Instead of mass-producing uric acid, the body safely accumulates the precursors hypoxanthine and xanthine. Because these specific precursors are significantly more water-soluble than uric acid, they are easily cleared by the kidneys.
Slide 12: Severe Defects in Nucleotide Degradation: Lesch-Nyhan Syndrome (LNS)

A single missing enzyme can swiftly transform molecular recycling into a complete neurological nightmare. This slide introduces one of the most severe inborn errors of metabolism, well known in medical science.
While standard gout is incredibly painful, true genetic failures in nucleotide degradation can be utterly devastating. Lesch-Nyhan Syndrome is a very rare, profoundly severe, X-linked recessive disorder caused by the complete absence of a crucial salvage enzyme: hypoxanthine-guanine phosphoribosyltransferase. Because it is strictly X-linked, this profound metabolic error occurs almost exclusively in male patients.
The full clinical presentation of this disastrous defect in nucleotide degradation is truly heartbreaking. Neurologically, patients miserably suffer from severe dystonia, resulting in highly involuntary muscle twisting, alongside significant developmental disabilities. Most uniquely, Lesch-Nyhan Syndrome causes a striking, completely irresistible urge for compulsive self-mutilation, frequently leading small patients to brutally bite off their own lips and fingers.
Biochemically, the absolute failure to successfully salvage bases means everything is incorrectly shunted toward complete nucleotide degradation. This quickly presents in early childhood as extreme, unmanageable hyperuricemia. The massive uric acid load physically damages the delicate renal system, causing painful hematuria and agonizing kidney stones.
Slide 13: The Cascade of Chaos: Understanding Nucleotide Degradation in LNS

Why exactly does a broken cellular recycling bin cause the whole biochemical factory to tragically explode? This slide clearly explains the exact biochemical cascade that strictly links a single missing salvage enzyme to massive uric acid overproduction.
The fundamental metabolic failure in Lesch-Nyhan Syndrome beautifully illustrates the highly interconnected nature of cellular pathways. When the vital salvage pathway fails due to missing HGPRT, purine nucleotide degradation becomes completely unregulated. Hypoxanthine and guanine cannot be recycled, leading to a massive, highly toxic buildup of the activated sugar substrate, PRPP.
Normally, PRPP is consumed in large amounts and safely during the standard salvage process. Its rapid, unchecked accumulation is totally disastrous. Furthermore, because salvage is completely halted, the vital cellular levels of IMP and GMP unexpectedly plummet. This severe drop forcefully removes critical negative feedback inhibition, creating a true perfect storm in the landscape of nucleotide degradation.
This severe imbalance heavily hyperstimulates the regulatory enzyme glutamine-PRPP amidotransferase. De novo synthesis skyrockets completely out of control. Because the resulting new purines cannot be cleanly salvaged, they are relentlessly funneled directly down the pathway of nucleotide degradation, resulting in massive, unchecked uric acid production that starves the central nervous system of vital nucleotides.
Slide 14: Immunodeficiency and Nucleotide Degradation: ADA-Deficient SCID
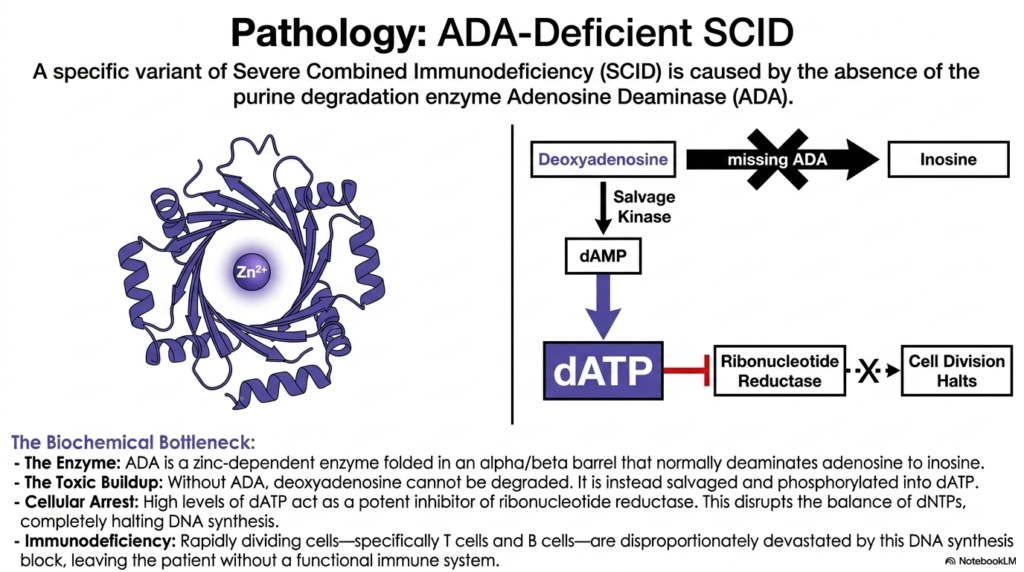
A small child forced to exist in a completely sterile bubble due to a missing immune system is actually suffering from a devastating metabolic traffic jam. This slide thoroughly explores how an invisible enzyme defect completely halts white blood cell division.
While Lesch-Nyhan Syndrome terribly affects the brain, another highly critical defect in purine nucleotide degradation completely devastates the developing immune system. A very specific variant of Severe Combined Immunodeficiency is directly caused by the absolute absence of Adenosine Deaminase. This unique zinc-dependent enzyme normally folds correctly into an alpha/beta barrel to safely deaminate adenosine into inosine.
Without Adenosine Deaminase, the normal, healthy flow of nucleotide degradation suddenly stalls at a catastrophic metabolic bottleneck. Deoxyadenosine simply cannot be properly degraded and safely removed from the cell. Instead, the trapped cell is wrongly forced to aggressively salvage the accumulating toxic buildup, rapidly phosphorylating deoxyadenosine into enormous, deadly levels of dATP.
These enormous cellular levels of dATP unexpectedly act as a highly potent, deadly inhibitor of ribonucleotide reductase, the exact enzyme responsible for safely making the biological precursors crucially needed for DNA synthesis. Rapidly dividing cells are disproportionately devastated by this specific failure of nucleotide degradation, ultimately leaving the young patient completely without any functional adaptive immunity.
Please read our Content Disclaimer Statement.
Check out our social media channels: