54. The Urea Cycle: Pathways of Nitrogen Metabolism
Imagine consuming a hearty, protein-rich meal, unaware that the resulting molecular breakdown products could rapidly damage the brain. The body constantly produces ammonia, a highly toxic byproduct of normal amino acid metabolism. This slide deck explores the fundamental biochemical pathways that successfully neutralize this daily threat. The core purpose of these slides is to guide medical and university students through the intricate enzymatic steps of nitrogen metabolism. By breaking down these complex structures, the presentation illuminates how the human liver elegantly safeguards the central nervous system from catastrophic toxicity.
Slide 1: Introduction to the Urea Cycle and Ammonia Detoxification
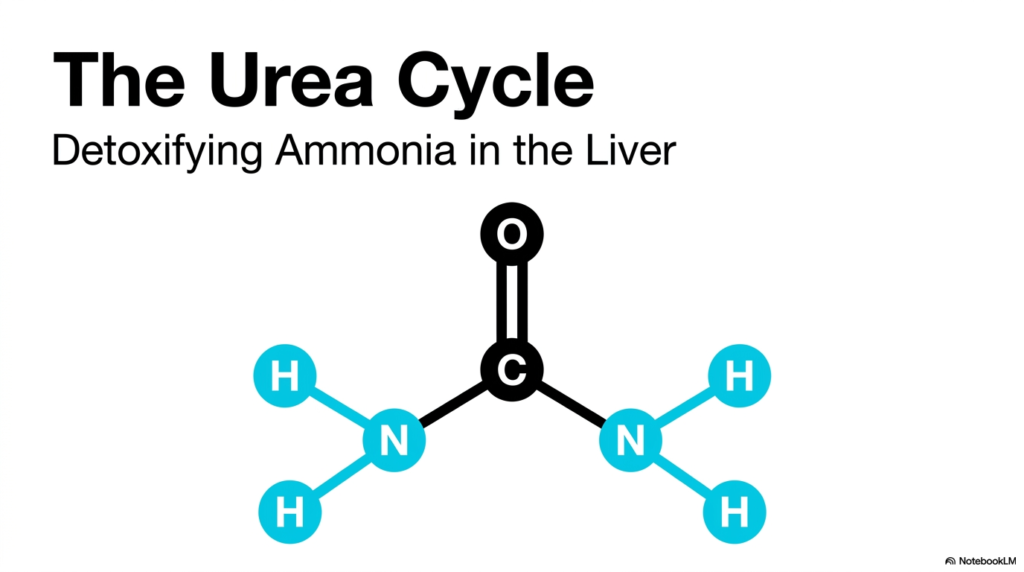
The visual on this opening slide introduces the primary chemical protagonist of the Urea Cycle: the urea molecule. At the center of the image lies a straightforward but highly significant biochemical structure, consisting of a central carbon atom double-bonded to an oxygen atom and single-bonded to two amino groups. This simple conformation represents the ultimate biochemical triumph of the liver. The primary objective of the Urea Cycle is the safe processing of volatile and dangerous nitrogenous waste into this remarkably stable, harmless compound. For medical students, recognizing this structural arrangement is the first step in understanding systemic nitrogen metabolism and its critical role in sustaining human life.
The transformation of toxic ammonia into this benign molecule does not happen by accident. The Urea Cycle is a carefully orchestrated sequence of biochemical events designed specifically for systemic detoxification. Without this metabolic pathway, the routine breakdown of dietary proteins would lead to a catastrophic accumulation of nitrogenous waste in the bloodstream. The slide sets the stage for a detailed exploration of hepatic function, emphasizing that the liver is the sole site of this crucial detoxification process. The elegant symmetry of the urea molecule belies the highly complex enzymatic machinery ultimately required to produce it.
As students progress through the remaining slides, they will discover how each functional group on this urea molecule is meticulously sourced and assembled. One amino group originates from free ammonia, while the other is donated by the amino acid aspartate. The central carbon and oxygen are derived directly from cellular bicarbonate. This structural breakdown illustrates the beauty of the Urea Cycle, demonstrating exactly how the body recycles and neutralizes cellular waste. Understanding this foundational concept prepares the learner for the deeper enzymatic intricacies that define human nitrogen metabolism and clinical biochemistry.
Ultimately, this title slide serves as a fundamental anchor for the complex biochemistry that follows. By visualizing the end product first, learners can better appreciate the intricate cellular gymnastics required to synthesize it. The liver bears an immense energetic burden in assembling this molecule, underscoring the evolutionary importance of preventing ammonia toxicity at all costs. This introductory overview ensures a solid conceptual foundation before diving into the individual enzymatic steps.
Slide 2: Evolutionary Context of the Urea Cycle and Ammonia Toxicity
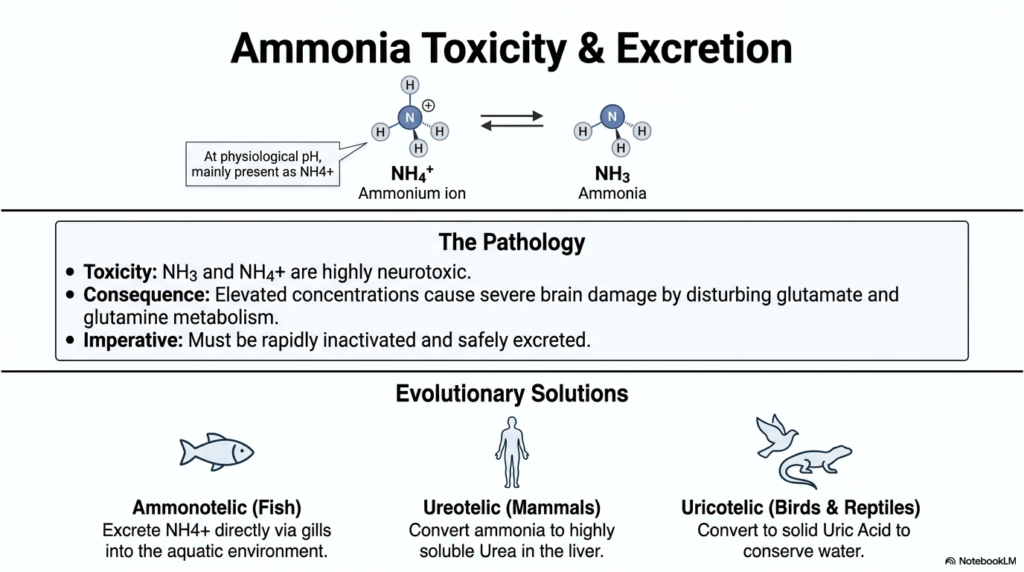
The pathology of unmanaged nitrogen is the primary focus of this slide, highlighting exactly why the Urea Cycle is an absolute non-negotiable requirement for mammalian survival. At physiological pH, ammonia exists primarily as the ammonium ion, though it constantly exists in equilibrium with free ammonia. Both chemical forms are highly neurotoxic. When the body fails to metabolize these compounds, elevated concentrations quickly traverse the blood-brain barrier. This infiltration wildly disturbs glutamate and glutamine metabolism in the brain, leading to severe neurological damage and potentially fatal swelling. The biological imperative is clear: the organism must rapidly inactivate and safely excrete this metabolic waste to maintain neurological integrity.
To solve this problem, nature has devised fascinating evolutionary strategies, with the mammalian Urea Cycle being just one highly prominent example. The slide categorizes organisms based on their specific nitrogen excretion methods. Aquatic animals, classified as ammonotelic, have the biological luxury of abundant water. Fish can simply excrete the ammonium ion directly through their gills into the surrounding aquatic environment, diluting the toxin instantly. This primitive yet highly effective method eliminates the need for complex internal metabolic conversions, provided the organism remains in a vast aqueous habitat.
Terrestrial animals face a completely different and more difficult environmental challenge. Mammals are ureotelic, meaning they rely exclusively on the Urea Cycle to convert toxic ammonia into highly soluble urea in the liver. This profound adaptation allows for the safe internal storage and transport of waste without requiring massive amounts of water for continuous, immediate dilution. Water conservation is absolutely crucial for terrestrial survival, enabling mammals to inhabit dry environments while safely managing daily protein catabolism and systemic nitrogen loads.
Finally, birds and reptiles represent the uricotelic evolutionary path. These highly adapted organisms convert ammonia into solid uric acid. This specific biochemical process requires even less water than mammalian excretion, making it ideal for egg-laying animals and those living in extreme, arid environmental conditions. By comparing these three distinct biological solutions, university students gain a profound appreciation for the specific environmental pressures that shaped human hepatic metabolism and the vital necessity of these intricate biochemical pathways.
Slide 3: Chemical Properties of the Urea Cycle End Product
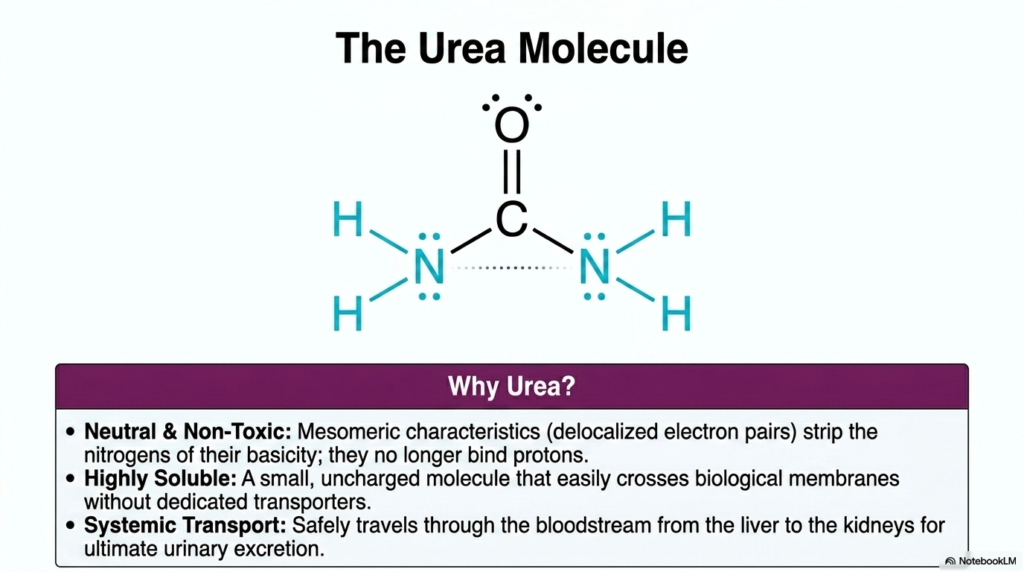
This slide delves into the molecular characteristics that make urea the ideal biological waste product for the Urea Cycle to generate. The chemical architecture of urea features mesomeric characteristics, meaning it contains highly delocalized electron pairs across its structure. This specific resonance structure effectively strips the nitrogen atoms of their typical, aggressive basicity. Consequently, these nitrogen atoms no longer actively bind ambient protons at physiological pH. Because of this protective electron delocalization, urea is exceptionally neutral and non-toxic, allowing it to safely accumulate in the bloodstream at much higher concentrations than free ammonia ever could.
Another critical biological feature highlighted here is the extreme aqueous solubility of urea. It is a very small, uncharged compound that dissolves readily in the aqueous environment of blood plasma. This immense systemic solubility is arguably one of the most important physiological outcomes of the entire Urea Cycle. Because it is uncharged and small, urea can easily traverse biological cell membranes without the need for dedicated, energy-consuming protein transporters. This passive diffusion significantly reduces the metabolic complexity required to physically move the waste product out of the liver cells and into the systemic circulation.
Systemic transport is the final piece of this vital metabolic puzzle. Once the liver has meticulously synthesized urea via the Urea Cycle, the stable molecule travels safely through the systemic bloodstream. It journeys smoothly from the hepatic system directly to the kidneys. In the renal system, it is rapidly filtered from the circulating blood and concentrated for final urinary excretion. This completely safe, non-toxic voyage depends entirely on the specific chemical and physical properties outlined in this slide.
For medical trainees, understanding the mesomeric properties of urea is not just an academic exercise in organic chemistry. It actively explains the fundamental, overarching rationale behind the liver’s metabolic design. If the final waste product were bulky, electrically charged, or chemically reactive, the human body would require entirely different, likely far more energetically expensive, mechanisms for safe transportation and elimination. The elegant simplicity of the final product demonstrates the remarkable efficiency of human biochemistry.
Slide 4: Cellular Architecture and Location of the Urea Cycle
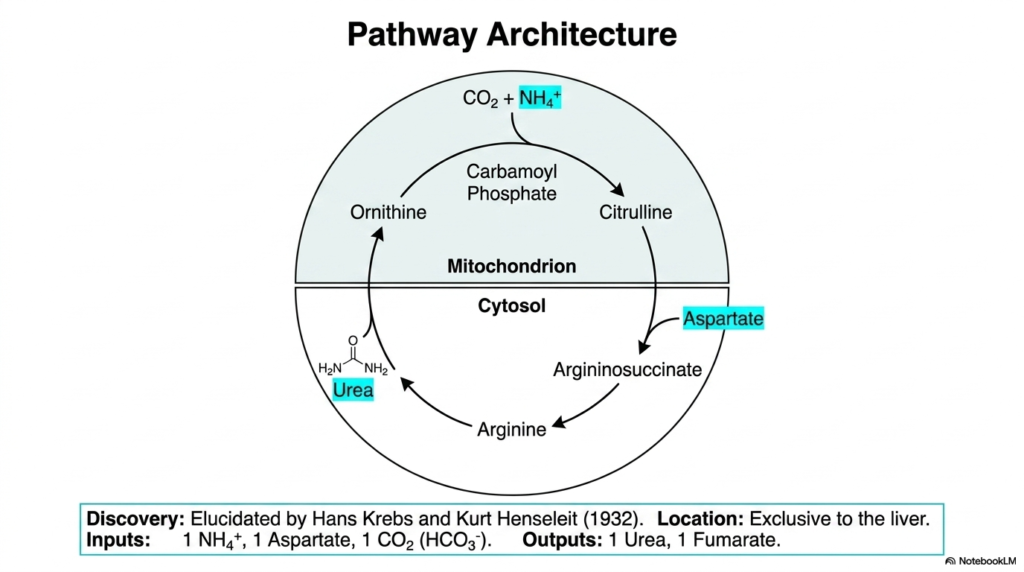
The pathway architecture shown here accurately maps the remarkable spatial organization of the Urea Cycle. Elucidated originally by Hans Krebs and Kurt Henseleit in 1932, this vital pathway is anatomically exclusive to the liver. A fascinating aspect of this metabolic process is its strict, non-negotiable compartmentalization within the hepatocyte. The cycle is deliberately divided between the mitochondrial matrix and the cellular cytosol. This precise spatial division prevents crucial chemical intermediates from wandering into competing metabolic pathways, ensuring that the critical detoxification of ammonia proceeds efficiently and without interruption. Students must memorize this dual-location feature, as it is a frequent topic in board examinations and a key factor in understanding metabolic regulation.
The initial steps of the Urea Cycle occur securely inside the protective mitochondrion. Here, raw metabolic inputs such as the ammonium ion and carbon dioxide are converted under enzymatic control into carbamoyl phosphate. This highly reactive intermediate then combines with ornithine to produce citrulline. The mitochondrion provides a highly specialized, tightly controlled environment for these volatile early reactions. By carefully sequestering free ammonia strictly within the mitochondrial matrix, the cell protects the delicate cytosolic components from unintended toxic side reactions, underscoring the protective evolutionary design of the cellular architecture.
Once the citrulline intermediate is safely formed, the biochemical action shifts dramatically across the mitochondrial membrane. Citrulline is exported directly into the cytosol, where the entire remainder of the Urea Cycle takes place. In this aqueous cellular fluid, aspartate donates the second essential nitrogen atom, officially forming argininosuccinate. This large intermediate is subsequently cleaved into arginine, which is finally hydrolyzed to release urea. The cycle is completed when the remaining ornithine backbone is actively transported back into the mitochondrion, ready to accept another carbamoyl group and begin the process anew.
This dual-compartment biological system prominently highlights the complex logistical requirements of nitrogen metabolism. Specific transport proteins heavily embedded in the inner mitochondrial membrane are absolutely vital for this operation, as they must continuously shuttle citrulline out and ornithine in. A genetic defect in these cellular transporters would immediately halt the entire metabolic sequence, clearly demonstrating that the physical location and spatial movement of these molecules are just as critical as the enzymatic reactions themselves.
Slide 5: Initiating the Urea Cycle with Carbamoyl Phosphate
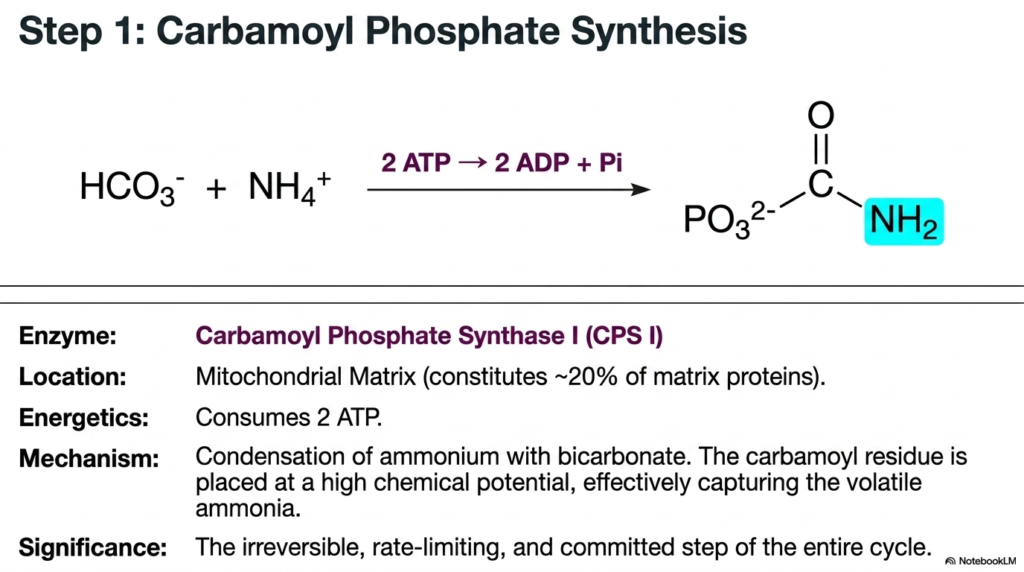
This slide introduces the absolute most critical and highly regulated step of the entire Urea Cycle: the synthesis of carbamoyl phosphate. The chemical reaction occurs deep within the mitochondrial matrix, using the enzyme Carbamoyl Phosphate Synthase I. This specific enzyme is remarkably abundant, constituting approximately 20% of the total protein content in the mitochondrial matrix. This immense cellular concentration clearly reflects the liver’s strong metabolic priority in capturing volatile ammonia before it can escape into the systemic circulation and cause catastrophic neurological damage to the host.
The complex chemical mechanism involves the direct condensation of an ammonium ion with cellular bicarbonate. However, creating this specific bonding arrangement requires a massive input of cellular energy. The reaction eagerly consumes two distinct ATP molecules, which are irreversibly hydrolyzed into two ADP molecules and inorganic phosphate. This incredibly heavy energetic investment places the resulting carbamoyl residue at an exceptionally high chemical potential. By expending this vital ATP, the Urea Cycle effectively traps the toxic nitrogen in a high-energy state, ensuring it is perfectly ready for the subsequent transfer to ornithine. The energetic cost here highlights the body’s willingness to sacrifice valuable ATP to prevent the disastrous consequences of ammonia accumulation.
From a biochemical regulatory standpoint, this is the singular defining moment of the Urea Cycle. The initial formation of carbamoyl phosphate by Carbamoyl Phosphate Synthase I is the strictly irreversible, rate-limiting, and completely committed step of the entire pathway. Once the targeted nitrogen is fully incorporated into this high-energy molecule, it is irreversibly destined for urinary excretion. The strict irreversibility perfectly prevents the highly dangerous backflow of free ammonia, absolutely guaranteeing that the biochemical momentum moves strictly forward toward safe, permanent elimination.
Medical students must focus heavily on the substantial energetic cost and the highly committed nature of this specific enzyme. Because it completely sets the biological pace for the entire metabolic sequence, any genetic deficiency or acquired impairment of Carbamoyl Phosphate Synthase I rapidly leads to the most severe forms of hyperammonemia. Understanding this very first enzymatic hurdle is absolutely essential for actively diagnosing and managing life-threatening metabolic crises in pediatric and adult patients alike.
Slide 6: Citrulline Formation in the Urea Cycle
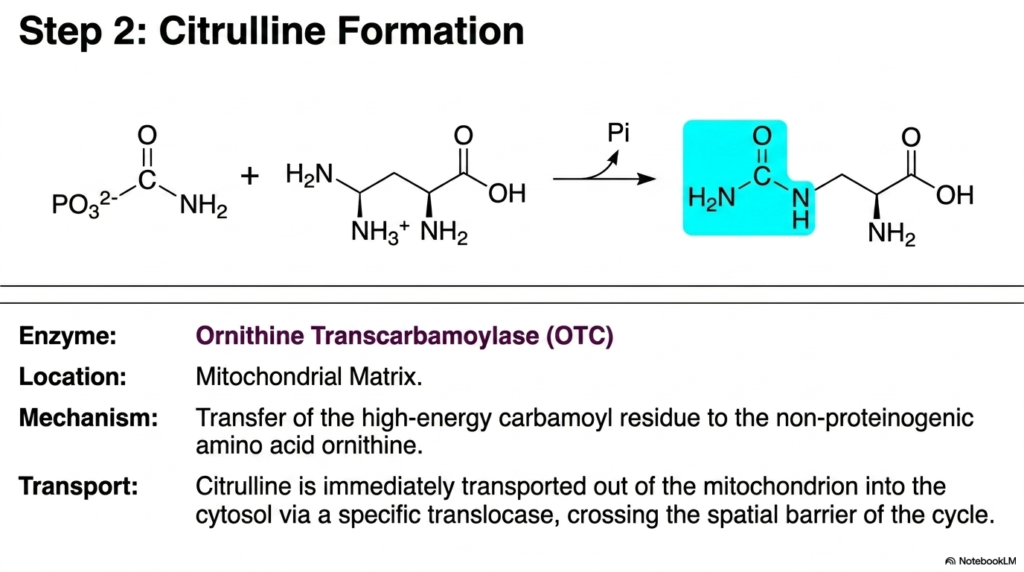
Following the deeply energetic capture of free ammonia, the Urea Cycle progresses immediately to its second crucial biochemical step: the formation of citrulline. This highly specific reaction is catalyzed by the enzyme Ornithine Transcarbamoylase, which also resides permanently within the highly protective environment of the mitochondrial matrix. The biochemical mechanism involves a rapid, precise transfer.
The high-energy carbamoyl residue, freshly synthesized in the very previous step, is transferred directly onto a waiting molecule of ornithine. Ornithine is a unique, non-proteinogenic amino acid that serves as a biological carrier molecule in this metabolic sequence. It acts as the structural foundation upon which the final waste product will be assembled, demonstrating the elegant recycling mechanisms inherent in cellular biology.
The vital formation of citrulline serves as a major metabolic bridge in the Urea Cycle. By securely attaching the volatile carbamoyl group to ornithine, the liver effectively creates a larger, slightly less reactive compound that is now fully prepared to physically leave the mitochondrion. As the high-energy bond in carbamoyl phosphate is rapidly broken during this transfer, inorganic phosphate is swiftly released. This crucial release of phosphate immediately provides the necessary thermodynamic driving force to ensure that the forward reaction proceeds rapidly and efficiently, thereby locking the first nitrogen atom securely into the growing organic framework.
A highly critical cellular logistical challenge immediately follows this enzymatic reaction. Citrulline is synthesized entirely within the mitochondrion, but the subsequent essential enzymes of the Urea Cycle are located strictly in the cellular cytosol. Therefore, citrulline must be transported out of the mitochondrial matrix immediately and efficiently. This spatial movement absolutely requires a highly specific translocase protein physically embedded in the inner mitochondrial membrane. This dedicated transporter allows the citrulline molecule to effortlessly cross the spatial barrier, smoothly shifting metabolic activity from the organelle into the main cellular fluid.
The immense clinical significance of this exact step simply cannot be overstated in academic biochemistry. Ornithine Transcarbamoylase is the most commonly mutated enzyme in inherited genetic nitrogen-processing disorders. When this vital enzyme fails, the cycle halts, preventing any citrulline formation and causing a rapid, deadly systemic buildup of ammonia and carbamoyl phosphate. For all future physicians, mastering the anatomical location and precise function of this enzyme is absolutely paramount to understanding the most common inherited defects encountered in clinical biochemistry.
Slide 7: Aspartate and the Urea Cycle Intermediates
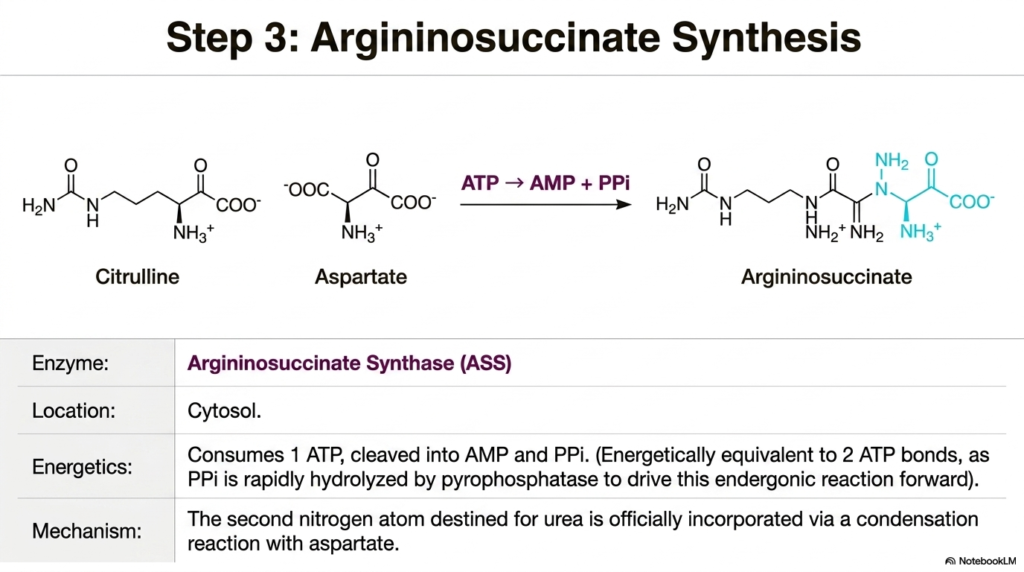
Step three introduces a truly monumental spatial shift in the Urea Cycle, as all the metabolic action has now fully relocated to the expansive cellular cytosol. It is exactly here that the highly anticipated second nitrogen atom, heavily destined for final excretion, is officially incorporated into the growing pathway. The vital enzyme solely responsible for this critical chemical addition is Argininosuccinate Synthase. This powerful enzyme carefully orchestrates a highly complex condensation reaction, physically binding the citrulline molecule that just exited the mitochondrion with a fresh molecule of aspartate. Aspartate serves brilliantly as the exclusive, dedicated nitrogen donor for this specific half of the final molecule’s structure.
This profound chemical condensation process is highly endergonic and requires a deeply significant energetic investment from the hepatocyte. The reaction heavily consumes one full molecule of cellular ATP. However, quite unlike the first step, this specific ATP is cleaved directly into adenosine monophosphate and inorganic pyrophosphate. This specific molecular cleavage pattern is thermodynamically equivalent to consuming two standard high-energy phosphate groups. The Urea Cycle boldly demands this steep energetic cost to ensure the irreversible union of these two large molecular structures directly against their natural energetic gradients.
The incredibly rapid cellular hydrolysis of the resulting pyrophosphate by the ubiquitous enzyme pyrophosphatase is what truly and powerfully drives this highly endergonic reaction strictly in the forward direction. By immediately and permanently destroying the pyrophosphate byproduct, the cell totally prevents the chemical reaction from ever running backward, flawlessly ensuring the strictly irreversible formation of argininosuccinate. This newly formed, incredibly bulky intermediate molecule now safely contains both of the targeted nitrogen atoms—one from the original free ammonia and one from aspartate—that will eventually constitute the final waste product of the Urea Cycle.
Deeply understanding the specific metabolic role of aspartate is incredibly crucial for properly grasping how the liver masterfully manages various internal pools of volatile nitrogen. The pathway absolutely does not solely rely on scavenging free ammonia; it actively and intelligently pulls dangerous amino groups from other metabolic circuits directly via aspartate. This vital step beautifully highlights the immense interconnectedness of cellular metabolism, clearly showing how the liver draws on distinct molecular resources to precisely assemble the final excretory compound efficiently and safely. Medical trainees must recognize Argininosuccinate Synthase as a major energetic toll bridge in the liver, reinforcing the concept that neutralizing neurotoxins is an incredibly costly cellular endeavor.
Slide 8: Argininosuccinate Cleavage in the Urea Cycle
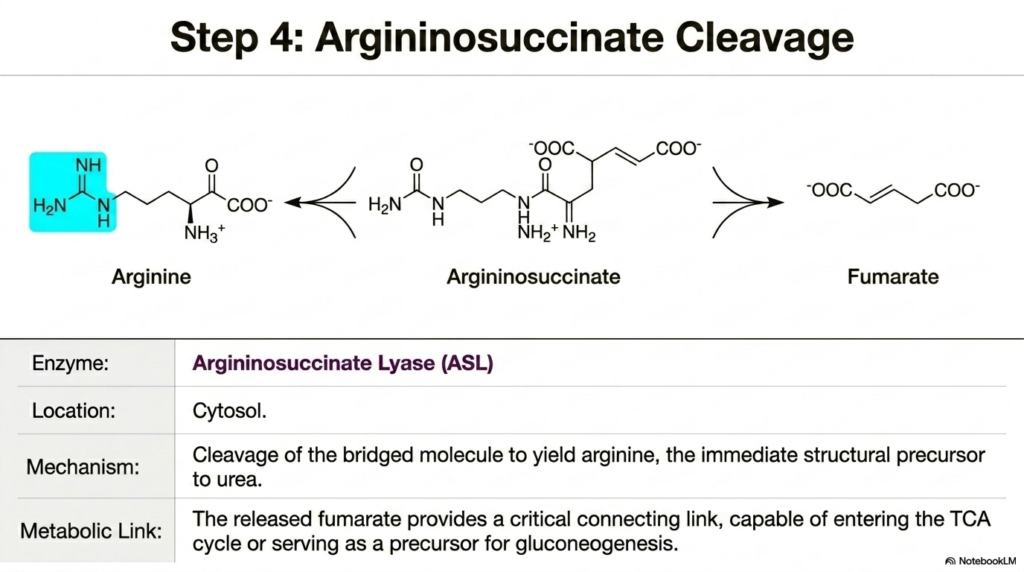
With the massive argininosuccinate molecule now fully and securely assembled, the Urea Cycle must finally begin trimming the heavy structure down to its final, functional excretory components. Step four introduces the highly crucial cleavage of this heavily bridged intermediate molecule. The fascinating enzyme Argininosuccinate Lyase acts decisively upon the complex, splitting it to yield two distinct chemical products: arginine and fumarate.
This precise enzymatic division is a truly magnificent biological example of molecular conservation, as the structural carbon backbone of aspartate is cleanly released while its deeply important donated nitrogen remains firmly attached to the core pathway. The cytosolic location of this event ensures that the newly generated arginine is perfectly positioned for the subsequent enzymatic hydrolysis.
The cellular formation of arginine boldly represents a highly significant metabolic milestone in the Urea Cycle. Arginine safely serves as the immediate, completely direct structural precursor to the final chemical waste product. It successfully and safely carries the entire guanidinium group intact, which securely contains both highly targeted nitrogen atoms that the human liver desperately seeks to quickly eliminate. For university students of biochemistry, correctly recognizing arginine not just as a standard dietary amino acid but as the final holding vessel for highly toxic nitrogen is the fundamental key to truly mastering hepatic metabolism.
Just as incredibly important as the arginine product is the rapid cellular release of the byproduct fumarate. The timely liberation of fumarate gracefully provides a critical metabolic link between entirely distinct cellular systems. Instead of being wasted, this fumarate can rapidly enter the tricarboxylic acid cycle to generate cellular energy, or it can readily serve as a vital carbon precursor for gluconeogenesis. This beautifully illustrates how the Urea Cycle is deeply and seamlessly integrated into the broader energetic and biosynthetic goals of the human body.
The precise molecular action of Argininosuccinate Lyase clearly demonstrates that clinical biochemistry is rarely ever a simple, purely linear, isolated process. The splitting of argininosuccinate is an incredibly efficient metabolic intersection. It vigorously pushes the deeply toxic nitrogen rapidly closer to its final exit while simultaneously and elegantly returning highly valuable carbon skeletons right back to the bustling cellular economy. Genetic deficiencies in this highly specific lyase result in argininosuccinic aciduria, an incredibly dangerous accumulation of the uncleaved intermediate that violently disrupts both systemic nitrogen disposal and overall energy balance.
Slide 9: Final Formation of Urea in the Urea Cycle
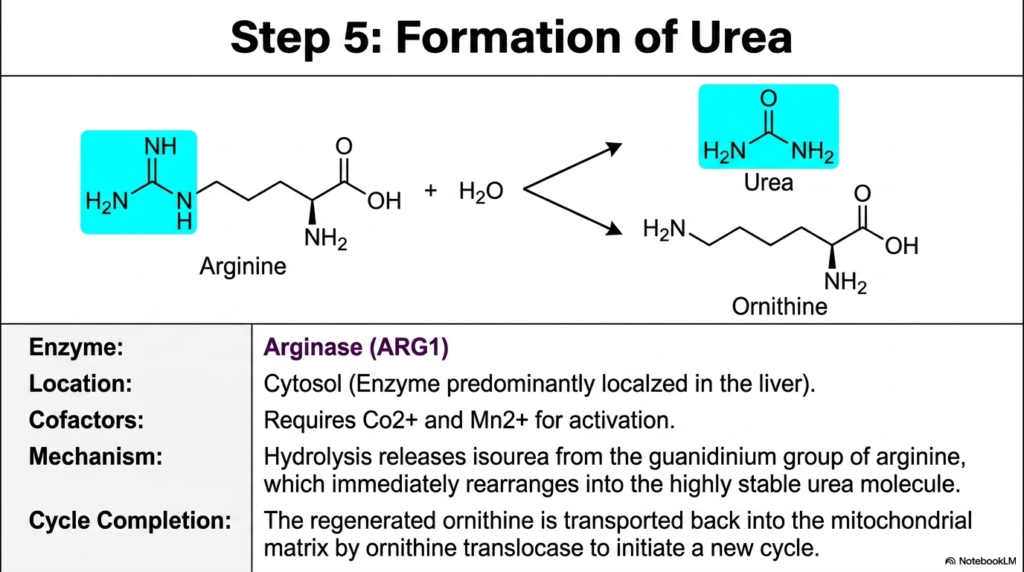
The deeply complex biochemical journey successfully culminates in step five, where the Urea Cycle finally and triumphantly achieves its ultimate systemic objective. The highly specialized enzyme Arginase, strictly localized within the hepatic cytosol, takes center stage. This powerful, dedicated enzyme flawlessly performs a highly targeted hydrolysis reaction, cleanly cleaving the exact arginine molecule safely produced in the very previous step.
Hydrolysis specifically and rapidly releases isourea directly from the guanidinium group of the arginine backbone. Almost instantaneously, this chemical intermediate permanently rearranges itself into the incredibly highly stable, perfectly symmetric urea molecule that the body can safely excrete. This decisive cut separates the dangerous nitrogen from the valuable amino acid skeleton, representing the crowning achievement of hepatic metabolic evolution and safeguarding the organism’s delicate nervous tissues.
Arginase is a deeply fascinating enzyme that strictly and exclusively requires highly specific metal cofactors for its activation, namely divalent cobalt and manganese ions. Without exactly these vital dietary micronutrients, the highly anticipated final cleavage absolutely cannot ever occur. This absolute dependency brilliantly highlights how trace minerals play a profoundly important role in essential macroscopic bodily functions, such as heavy detoxification. Once the Urea Cycle triumphantly produces this final urea molecule, the highly stable compound simply diffuses out of the hepatocyte, enters the systemic bloodstream, and eagerly begins its entirely harmless transit toward the renal filtration system.
The sheer metabolic genius of the Urea Cycle lies in its flawlessly cyclic nature, which is beautifully and precisely demonstrated at this final, highly exact juncture. When cellular arginase hydrolyzes arginine to release the vital waste product, the remaining structural backbone is not discarded. Instead, the precise enzymatic cleavage beautifully regenerates a pristine ornithine molecule. This exact ornithine molecule is then immediately and efficiently transported right back across the heavily guarded mitochondrial membrane strictly by the dedicated ornithine translocase protein, fully ready to joyfully accept a brand-new carbamoyl group and properly initiate a completely brand-new cycle.
This continuous, highly rapid regeneration of ornithine is exactly what boldly makes the pathway a truly functional biological cycle rather than a simple, highly wasteful linear track. By constantly and beautifully recycling the crucial carrier molecule, the heavily burdened liver drastically and successfully minimizes the metabolic resources required to maintain a constant level of systemic nitrogen disposal. University students must deeply appreciate this specific step not merely as the final end of the line for the dangerous waste, but rather as the mandatory biochemical reset button that enables the liver to rapidly process continuous waves of heavy dietary protein.
Slide 10: Overall Energetics of the Urea Cycle

This important slide cautiously steps back to examine the broad, macroscopic accounting of the Urea Cycle, focusing heavily on the steep energetic costs strictly required to continuously maintain a perfectly non-toxic internal cellular environment. The overall balanced metabolic reaction clearly reveals that safely and securely packaging two separate molecules of highly toxic ammonia immediately alongside one molecule of carbon dioxide fiercely demands a massive cellular investment.
Specifically, the entire pathway vigorously consumes three full ATP molecules, but, strictly due to the aggressive pyrophosphate cleavage in step three, the actual net cost is precisely four high-energy phosphate groups to synthesize just one single, isolated molecule of urea. For medical students, this accounting is crucial; it demonstrates that ammonia detoxification is prioritized over almost all other cellular functions, thereby hoarding ATP to prevent rapid neurological decline.
This remarkably heavy energy consumption beautifully represents the absolutely true biological cost of doing regular physiological business as a complex terrestrial organism. The initial robust capture of free ammonia directly by Carbamoyl Phosphate Synthase I greedily consumes two ATP, while Argininosuccinate Synthase subsequently heavily consumes the energetic equivalent of precisely two more. The Urea Cycle is thus a deeply endergonic pathway, meaning it simply cannot run freely without a fiercely constant, truly massive continuous influx of valuable cellular energy. The human liver pays a steep, continuous metabolic tax to ensure the fragile nervous system remains perfectly shielded from volatile systemic nitrogen.
However, complex cellular biochemistry is also remarkably and deeply economical, and the Urea Cycle beautifully features a truly brilliant, highly effective energetic offset mechanism. The crucial metabolic key smoothly lies entirely in the specific fumarate molecule, eagerly released during step four. Out in the vast cytosol, this specific fumarate is rapidly hydrated to form malate, which is then rapidly oxidized to oxaloacetate. This specific, highly important cytosolic oxidation reaction beautifully and efficiently generates exactly one full molecule of valuable cytosolic NADH. This is an absolutely crucial energetic recovery step, seamlessly allowing the burdened cell to slightly recoup some of the truly massive energetic losses severely incurred strictly during the demanding earlier stages.
Through the vital action of the cellular malate-aspartate shuttle, this highly valuable single molecule of newly formed NADH is smoothly transferred completely to the mitochondrial electron transport chain, where it ultimately seamlessly yields approximately two and a half ATP strictly through robust oxidative phosphorylation. Therefore, while the initial gross metabolic energetic cost is a heavy four high-energy bonds, the smooth recovery of this exact NADH significantly and beautifully mitigates the pathway’s deeply steep metabolic toll. Deeply understanding this exact biochemical balance is absolutely vital for fully comprehending how the robust liver effortlessly manages massive energy budgets during periods of truly high, continuous dietary protein turnover.
Slide 11: Allosteric Regulation of the Urea Cycle
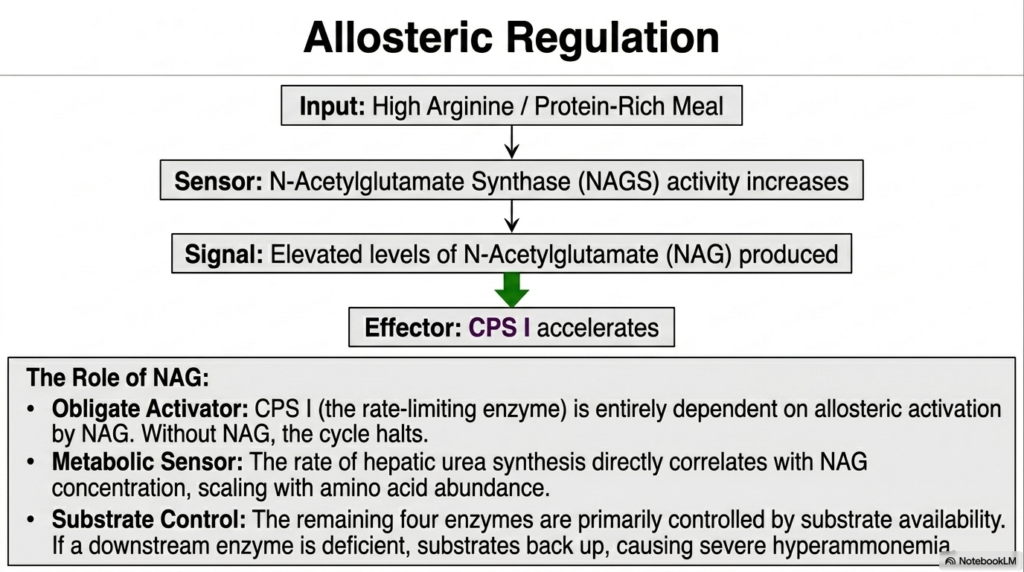
Complex metabolic pathways absolutely must adapt smoothly to shifting environmental inputs, and the Urea Cycle is exquisitely and highly tightly regulated strictly to elegantly match the body’s fluctuating dietary protein intake. The crucial primary input signal for this elegant regulatory system is a high concentration of the dietary amino acid arginine, typically found strictly following a hearty, protein-rich meal.
This sheer abundance beautifully acts as a sensitive biochemical sensor, completely and directly increasing the vigorous activity of a vital enzyme properly called N-Acetylglutamate Synthase. This specific enzyme’s sole metabolic purpose is to rapidly produce highly elevated intracellular levels of a highly specialized signaling molecule, broadly known as N-Acetylglutamate (NAG).
The absolutely critical role of NAG is strictly within the bounds of the Urea Cycle and is completely non-negotiable. It serves perfectly as the obligate allosteric activator for Carbamoyl Phosphate Synthase I, the profoundly important rate-limiting enzyme, heavily discussed strictly in the early steps. Without the highly specific physical binding of NAG completely to this exact enzyme, the entire massive cycle strictly halts instantly.
This smoothly creates a truly brilliant, beautifully dynamic cellular feedback loop. When systemic protein intake is extremely high, cellular amino acid abundance rapidly scales up vital NAG production, which in turn forcefully drives the crucial rate-limiting enzyme into its fully active conformation. This ensures that the liver expends its massive ATP reserves on ammonia detoxification only when there is a physiological need to process incoming dietary or catabolic nitrogen loads.
Because of this deeply elegant mechanism, the true rate of vital hepatic urea synthesis correlates exactly and directly with the precise intracellular NAG concentration. It elegantly operates perfectly as a highly sensitive cellular metabolic throttle. When the human liver suddenly faces a massive flood of incoming amino groups, the cellular throttle powerfully opens extremely wide, aggressively accelerating the entire Urea Cycle to rapidly clear the dangerous sudden influx of highly toxic ammonia. Conversely, during deep starvation or extremely low-protein diets, cellular NAG levels plummet smoothly and rapidly, thereby slowing the entire cycle to conserve highly valuable ATP and vital carbon skeletons.
Beyond this deeply primary, highly specific allosteric control, the remaining four enzymes of the pathway are primarily controlled by simple substrate availability. They strictly process absolutely whatever exact chemical intermediates are physically handed directly to them. This clearly means that if a deeply vital downstream enzyme is utterly genetically deficient, vital substrates will violently and catastrophically back completely up the entire chain, utterly overwhelming the delicate, highly precise regulatory balance and rapidly causing severe hyperammonemia regardless of internal NAG levels. Truly understanding the precise NAG function is essential for fully mastering highly targeted pharmacological interventions strictly in absolutely certain catastrophic metabolic crises.
Slide 12: Integration via the Krebs Bicycles in the Urea Cycle
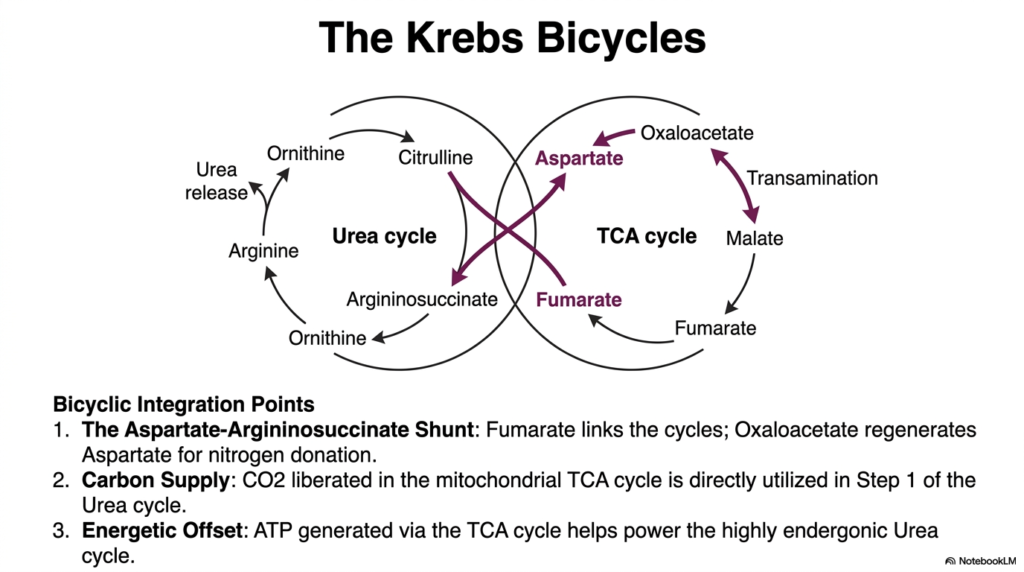
This beautiful final schematic slide illustrates a highly elegant concept strictly in deep cellular biochemistry, widely known as the Krebs Cycle, seamlessly demonstrating exactly how the entire Urea Cycle absolutely does not operate quietly in a purely lonely metabolic vacuum. Instead, it is highly physically and deeply chemically intertwined with the massive Tricarboxylic Acid cycle.
The most prominent metabolic integration point is definitely the highly vital aspartate-argininosuccinate shunt. Fumarate, released exclusively during the heavy cleavage of argininosuccinate, rapidly serves as the primary metabolic bridge, linking these two vital cellular engines and allowing carbon and nitrogen to flow between them seamlessly. This intricate biochemical choreography highlights how the body maximizes the utility of every intermediate molecule, preventing metabolic bottlenecks and ensuring continuous flux through both systems.
As this highly vital fumarate smoothly enters the bustling energetic cellular pathways, it is eventually flawlessly converted into oxaloacetate. Through the seamless, highly efficient process of rapid transamination, this vital oxaloacetate is immediately and completely utilized to regenerate fresh aspartate. This extremely newly formed fresh aspartate is then effortlessly funneled strictly right completely back straight into the bustling Urea Cycle completely to proudly serve immediately as a primary dedicated nitrogen donor heavily once again.
This massive, truly amazing bicyclic cellular intersection seamlessly ensures that the absolutely essential, highly required, vital nitrogen carriers are continuously and deeply recycled and fully available, completely preventing the busy liver from ever severely stalling due to a massive shortage of vital donor molecules.
Furthermore, the incredible Krebs Bicycles beautifully and effortlessly provide a highly direct, entirely continuous vital carbon supply. The raw carbon dioxide heavily liberated constantly strictly during the deeply robust mitochondrial oxidative cellular processes is immediately and directly captured beautifully and highly utilized strictly in the absolutely very first exact step strictly of the entire Urea Cycle completely to form completely vital carbamoyl phosphate.
The exact heavy exhaust rapidly exiting from one massive cellular engine is truly immediately completely fully recycled rapidly strictly as the exact raw foundational material strictly for entirely another. This deeply, absolutely, profoundly beautiful integration showcases the highly efficient, highly beautiful hepatocyte, leaving absolutely virtually completely no single molecular cellular waste unutilized.
Finally, this deep cellular integration powerfully reinforces the highly crucial energetic offset, which was fully discussed previously. The vital ATP is constantly generated strictly via the robust oxidative actions of the major adjacent biological pathways, which actively and seamlessly help intensely power the highly endergonic systemic nitrogen disposal process. For dedicated university students, carefully visualizing these highly overlapping complex circles is entirely critical.
It completely smoothly totally transforms the mere rote memorization entirely of completely isolated biological pathways beautifully deeply into a remarkably deep, truly incredibly holistic total understanding entirely of human cellular biology, absolutely firmly completely proving that energetic active metabolism and crucial nitrogen-heavy detoxification absolutely strictly are definitely just two exact deeply interconnected sides entirely of the truly perfectly exact same profound biological coin.
Slide 13: Diagnosing Urea Cycle Disorders
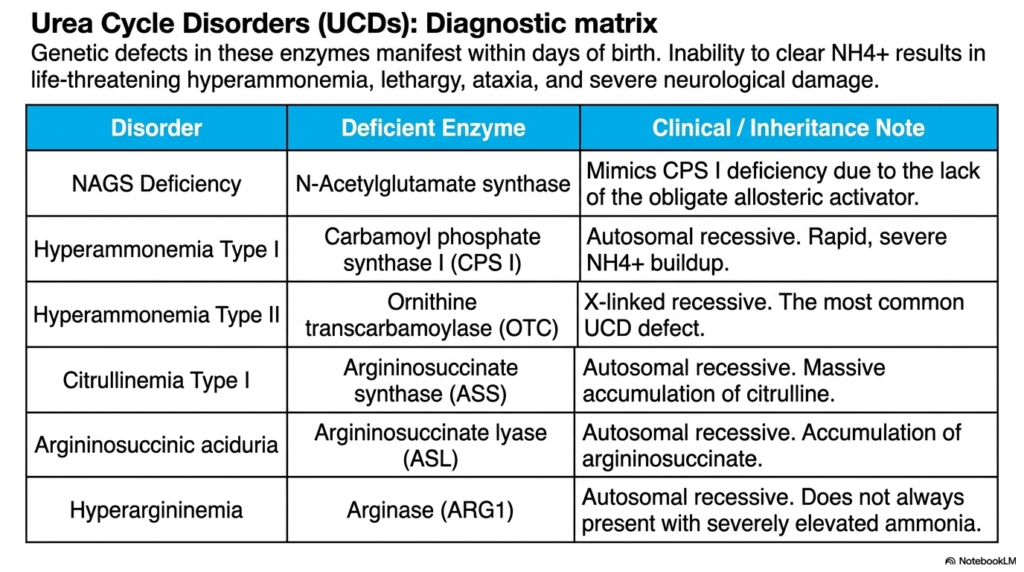
The final slide presents a critical diagnostic matrix outlining the severe clinical consequences of genetic defects in the urea cycle. Known collectively as Urea Cycle Disorders, or UCDs, these enzymatic failures typically manifest within mere days of birth. When an infant inherits a defective enzyme, their liver is unable to clear the ammonium ion. This rapid biochemical failure results in life-threatening hyperammonemia, characterized clinically by profound lethargy, severe ataxia, and devastating, irreversible neurological damage if not immediately corrected. Because the toxic burden accumulates so swiftly after the neonate’s first protein feedings, rapid recognition of these enzymatic blockades is a fundamental competency for every practicing pediatrician and metabolic geneticist.
The matrix breaks down each specific disorder by its deficient enzyme. A deficiency in N-Acetylglutamate Synthase mimics a Type I defect precisely because it eliminates the obligate allosteric activator required to start the Urea Cycle. Hyperammonemia Type I involves a direct defect in the rate-limiting enzyme, Carbamoyl Phosphate Synthase I, leading to a catastrophic, rapid buildup of toxins. Both of these proximal defects are inherited in an autosomal recessive manner and present with extreme, sudden neurological crises.
Interestingly, Hyperammonemia Type II, caused by a defect in Ornithine Transcarbamoylase, is an X-linked recessive disorder. It is the most common single defect encountered in clinical practice. Further down the pathway, Citrullinemia Type I and Argininosuccinic aciduria cause massive cellular accumulations of citrulline and argininosuccinate, respectively. These distal defects still severely impair the Urea Cycle but offer different diagnostic biochemical markers in the blood and urine due to the specific intermediate that backs up.
Finally, Hyperargininemia, caused by an arginase deficiency, is unique because it does not always present with the severely elevated ammonia levels seen in the other disorders. Instead, progressive spasticity and developmental delay are common. For medical professionals, memorizing this precise matrix is non-negotiable. Pinpointing the exact enzymatic failure dictates the specific dietary restrictions, nitrogen-scavenging medications, and long-term management strategies required to save a newborn’s life and preserve their cognitive function.
Please read our Content Disclaimer Statement.
Check out our social media channels: