53. Amino Acid Degradation: A Biochemical Analysis
Welcome to this comprehensive breakdown of protein and nitrogen metabolism. The following explanations accompany the slide deck series, providing an in-depth academic exploration of these complex biochemical pathways for college and medical students.
Slide 1: Amino Acid Degradation: The Big Picture of Protein Metabolism
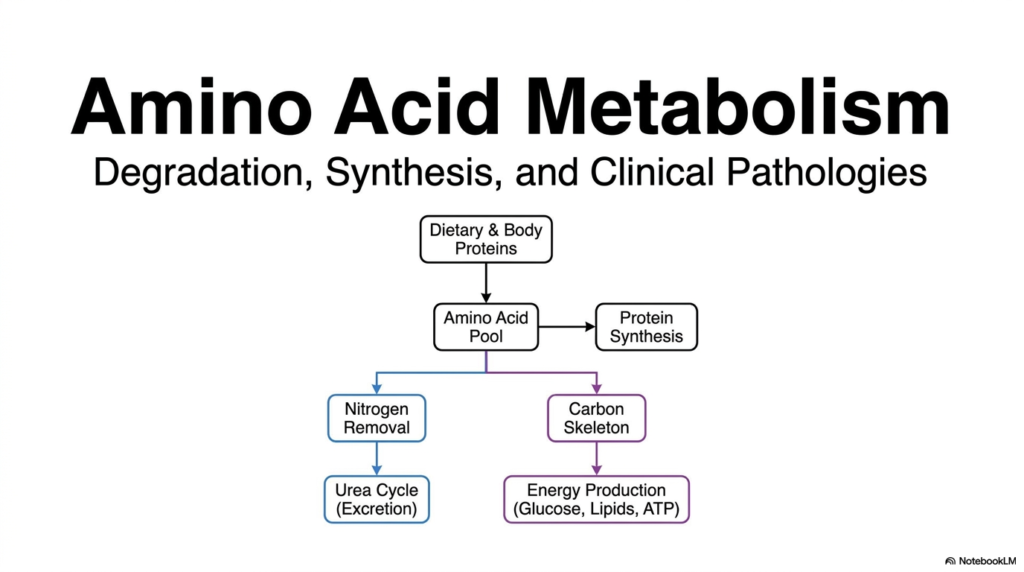
Students and researchers often marvel at how the human body perfectly balances the constant breakdown and building of biological tissues. This delicate physiological balance relies heavily on the complex metabolic pathways of Amino Acid Degradation. The core purpose of this slide is to introduce the overarching framework of how dietary and cellular proteins enter a central metabolic pool and are subsequently managed for sustained energy production.
When proteins are consumed through the diet or intracellular proteins are recycled, they contribute directly to a central metabolic reservoir. From this shared pool, these vital building blocks can be directed toward the synthesis of new proteins. However, biological systems cannot store excess quantities of these nitrogen-containing compounds. This is exactly where the vital process of Amino Acid Degradation becomes critical. The body must efficiently manage any surplus by splitting the molecule into two distinct parts: the nitrogen component and the carbon skeleton.
Understanding Amino Acid Degradation requires carefully tracking both components, as their biochemical fates diverge completely. The nitrogen group is inherently toxic if left to accumulate as free ammonia. Therefore, it must be safely channeled into the urea cycle for eventual renal excretion. This nitrogen removal is a non-negotiable requirement for cellular survival, specifically protecting the central nervous system from severe hyperammonemia and neurological damage.
Meanwhile, the remaining carbon skeleton is far from useless metabolic waste. It is seamlessly repurposed for cellular energy production. Depending on the body’s physiological needs, these carbon structures feed directly into metabolic pathways that generate glucose, synthesize lipids, or produce ATP. Through effective Amino Acid Degradation, biological systems ensure that no potential energy source is ever wasted, providing a fundamental roadmap for understanding both normal metabolism and severe clinical pathologies.
Slide 2: Amino Acid Degradation: The Lifecycle of Proteins
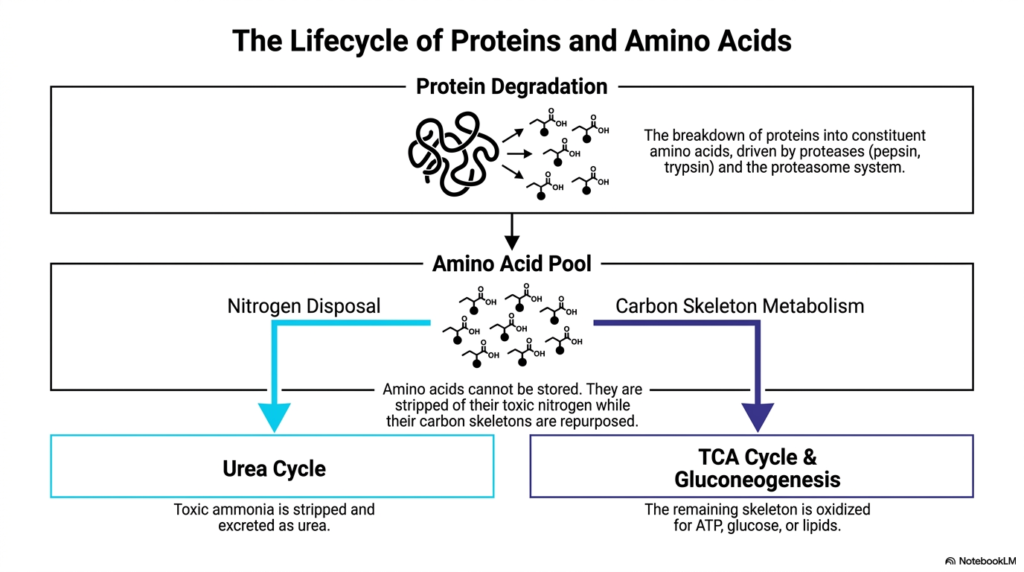
The journey from a complex folded protein to usable metabolic energy is a true marvel of cellular engineering and biochemistry. The core purpose of this slide is to visualize the complete lifecycle of proteins, detailing how macromolecules are systematically dismantled and routed through Amino Acid Degradation to sustain human life and maintain crucial energy homeostasis.
Protein degradation initiates this continuous cycle through the targeted enzymatic breakdown of long polypeptide chains into their constituent molecular parts. This digestive and recycling process is driven by specific proteases such as pepsin and trypsin in the gut, alongside the highly regulated intracellular proteasome system. Once thoroughly dismantled, these liberated molecules enter a shared metabolic pool, remaining temporarily available for either new protein synthesis or immediate Amino Acid Degradation.
Because biological systems entirely lack a dedicated storage mechanism for these nitrogenous compounds, any dietary or cellular excess must immediately undergo Amino Acid Degradation. This essential catabolic process physically strips the potentially toxic nitrogen group away from the energetically valuable carbon skeleton. The metabolic pathways diverge sharply at this exact juncture, representing a critical metabolic intersection for maintaining cellular homeostasis and preventing acute toxicity.
The nitrogen disposal pathway specifically funnels toxic ammonia directly into the Urea Cycle. Here, the liver safely converts it into highly soluble urea for rapid renal excretion. Conversely, the carbon skeleton metabolism pathway captures the remaining molecular structures. These carbon skeletons are heavily oxidized and routed directly into the TCA Cycle or utilized for Gluconeogenesis. Through the highly efficient mechanisms of Amino Acid Degradation, the human body transforms potential metabolic toxins into invaluable life-sustaining substrates, continuously yielding essential ATP, glucose, or lipids.
Slide 3: Amino Acid Degradation: Separating the Amino Group
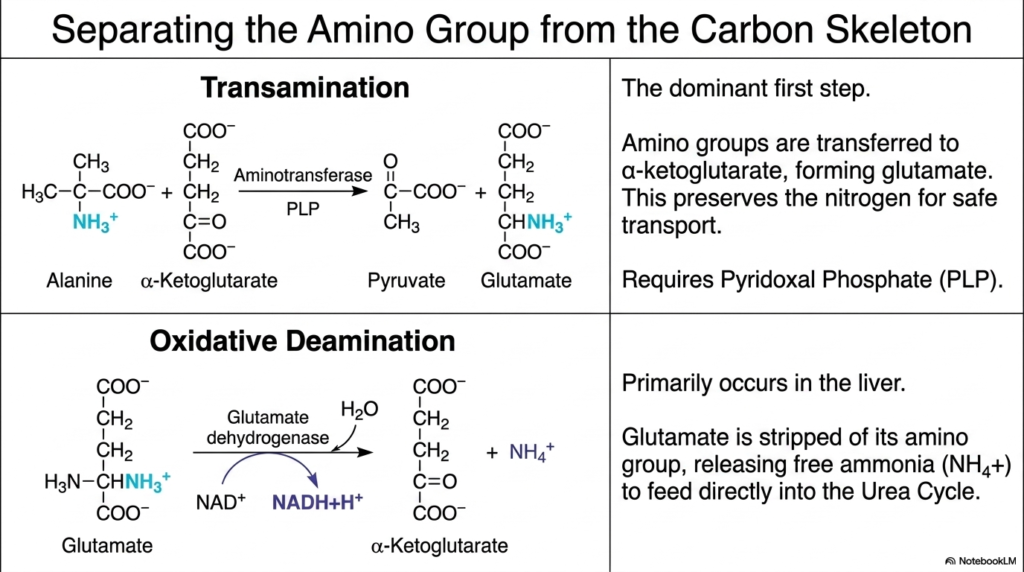
Biochemists recognize that handling volatile nitrogen is one of the most dangerous yet essential tasks a biological cell performs daily. The core purpose of this slide is to detail the precise enzymatic mechanisms of Amino Acid Degradation, specifically focusing on how the potentially toxic amino group is safely separated from the valuable carbon skeleton through transamination and oxidative deamination.
The first and dominant step in Amino Acid Degradation is transamination. During this crucial reaction, amino groups are carefully transferred from a donor amino acid to alpha-ketoglutarate. This enzymatic transfer creates glutamate, a molecule that safely preserves the nitrogen for secure cellular transport. This specific reaction is catalyzed by aminotransferases and strictly requires Pyridoxal Phosphate, commonly known as PLP or Vitamin B6, as an essential enzymatic cofactor for success.
Following transamination, oxidative deamination primarily occurs within the liver’s highly protective environment. Here, the newly formed glutamate is stripped of its amino group by the enzyme glutamate dehydrogenase. This decisive step in Amino Acid Degradation releases free ammonia directly into the surrounding cellular environment, while simultaneously generating valuable reducing equivalents in the form of NADH, which can later be harvested for ATP.
The free ammonia generated by oxidative deamination is highly toxic to cellular structures and must be swiftly managed by the body. It feeds directly into the Urea Cycle for safe detoxification and eventual excretion. Together, these two fundamental steps of Amino Acid Degradation ensure that the body can safely harvest the energy locked within carbon skeletons without suffering from catastrophic nitrogen toxicity or cellular death.
Slide 4: Amino Acid Degradation: Converging on 7 Intermediates
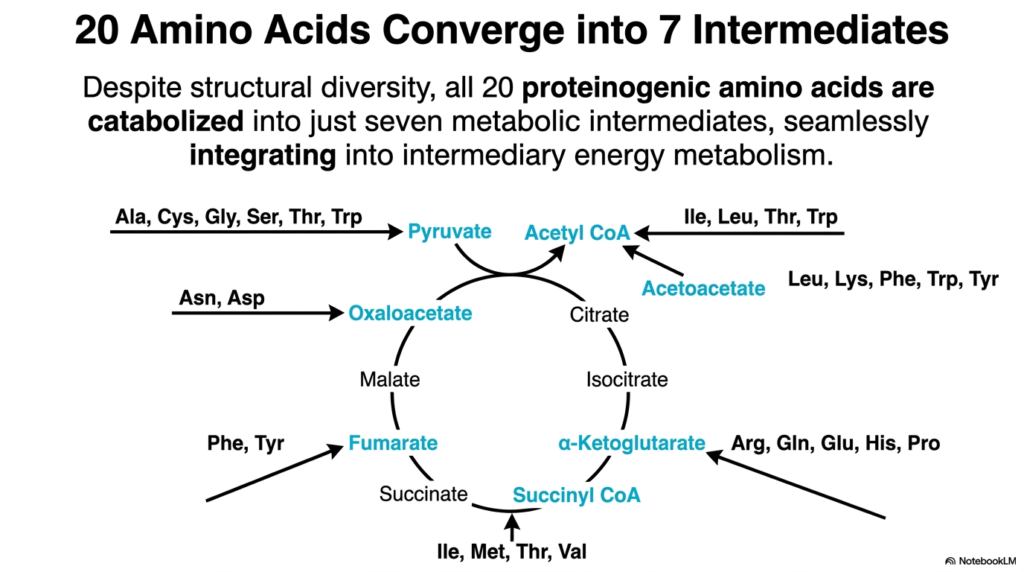
Metabolism is often compared to a complex highway system where diverse local roads eventually merge into a few major, high-speed superhighways. The core purpose of this slide is to demonstrate how the twenty structurally unique proteinogenic amino acids are systematically catabolized during Amino Acid Degradation into just seven central metabolic intermediates.
Despite the immense structural diversity found among the twenty standard amino acids, their catabolic pathways are incredibly streamlined and highly efficient. Through the various highly regulated enzymatic steps of Amino Acid Degradation, these complex molecules are systematically broken down and funneled into a concentrated set of metabolic end products. This elegant biological convergence seamlessly integrates complex protein catabolism directly into the body’s primary intermediary energy metabolism.
The seven resulting intermediates include pyruvate, acetyl CoA, acetoacetate, alpha-ketoglutarate, succinyl CoA, fumarate, and oxaloacetate. For instance, molecules like alanine, cysteine, and serine directly yield pyruvate, while others like aspartate efficiently yield oxaloacetate. This funneling process highlights the sheer efficiency of Amino Acid Degradation, allowing the body to use common metabolic machinery to process a wide variety of dietary inputs without needing endless, unique biochemical pathways.
Once these specific metabolic intermediates are formed, they can directly enter the Tricarboxylic Acid (TCA) cycle. This integration means that the final end products of Amino Acid Degradation are virtually indistinguishable from the energetic intermediates produced by carbohydrate or lipid metabolism. Ultimately, this convergent design provides the cellular machinery with maximum biochemical flexibility, ensuring that energy extraction remains robust and adaptable under shifting physiological conditions.
Slide 5: Amino Acid Degradation: Classification by Metabolic Potential
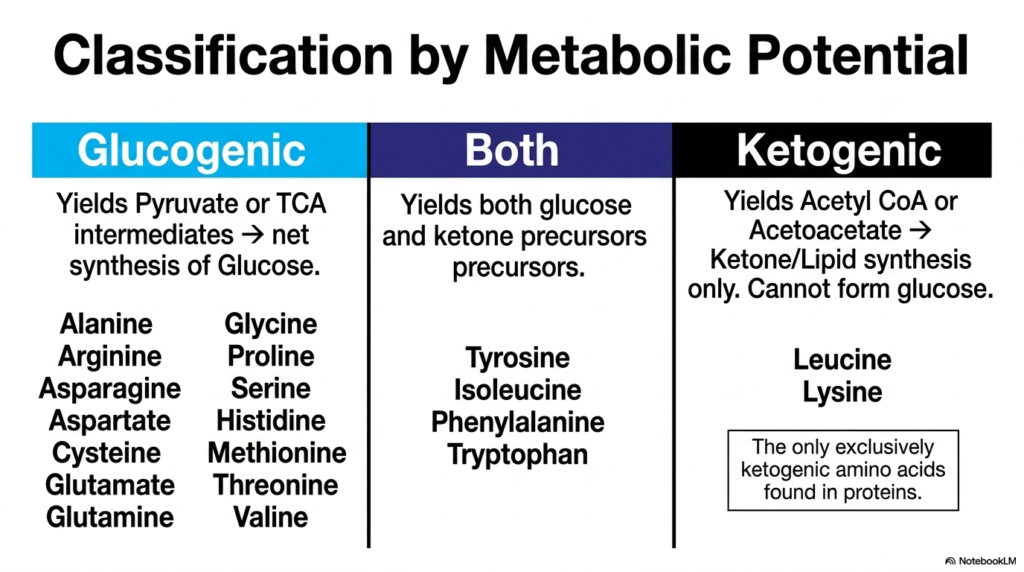
Medical students must frequently categorize metabolic substrates to accurately predict how the body will respond during states of prolonged fasting or acute starvation. The core purpose of this slide is to classify molecules undergoing Amino Acid Degradation based on their inherent metabolic potential, formally dividing them into glucogenic, ketogenic, or highly adaptable dual-purpose categories.
Glucogenic amino acids are those that specifically yield pyruvate or TCA cycle intermediates upon complete metabolic catabolism. This specific metabolic fate is crucially important because these intermediates can be directly channeled toward the net synthesis of new glucose via gluconeogenesis. During intense periods of fasting, the Amino Acid Degradation of glucogenic molecules such as alanine, serine, and glutamate provides the essential blood glucose required to sustain high brain function and red blood cell metabolism.
Conversely, ketogenic amino acids exclusively yield acetyl CoA or acetoacetate during their specific Amino Acid Degradation. These specific end products cannot be used to synthesize glucose; instead, they are strictly channeled into ketone body production or general lipid synthesis. Leucine and lysine hold a very unique physiological distinction here, as they are the only exclusively ketogenic amino acids found within human proteins, making them highly specialized metabolic fuels.
Finally, a select group of molecules, including tyrosine, isoleucine, phenylalanine, and tryptophan, exhibit remarkable dual metabolic capabilities. The Amino Acid Degradation of these complex molecular structures yields precursors for both glucose and ketone bodies simultaneously. Understanding these strict energetic classifications is absolutely vital for predicting metabolic outcomes in clinical scenarios, especially when managing severe dietary interventions or treating dangerous metabolic disorders.
Slide 6: Amino Acid Degradation: S-Adenosylmethionine and Methyl Transfer
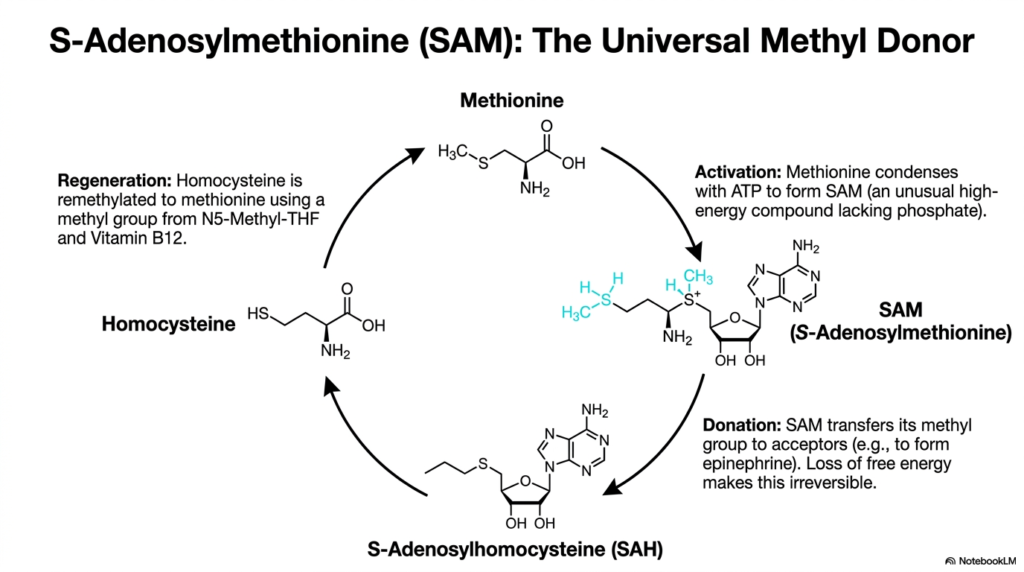
The transfer of single carbon units is a microscopic molecular event that has massive, body-wide implications for DNA regulation and neurotransmitter synthesis. The core purpose of this slide is to deeply explore the methionine cycle, a vital biochemical pathway running parallel to standard Amino Acid Degradation, focusing heavily on the creation and vital function of S-Adenosylmethionine.
Methionine metabolism is biochemically unique because it serves two critical roles: structural protein synthesis and essential cellular methyl donation. The activation phase prominently begins when methionine condenses with ATP to form S-Adenosylmethionine, commonly referred to as SAM. Interestingly, this reaction creates an unusual, high-energy compound that completely lacks a traditional phosphate group, yet possesses immense biochemical potential for complex downstream metabolic reactions.
Once formed, SAM acts as the universal methyl donor throughout human biochemistry. During the donation phase, SAM eagerly transfers its valuable methyl group to various acceptors, a process that is absolutely critical for synthesizing complex compounds such as epinephrine. This transfer involves a significant loss of free energy, rendering the reaction completely irreversible and ensuring that the overall metabolic cycle moves continuously forward alongside parallel Amino Acid Degradation.
After donating its methyl group, SAM is rapidly converted to S-Adenosylhomocysteine, which is subsequently hydrolyzed to homocysteine. From here, homocysteine must be remethylated back to methionine to sustain the cycle, a critical regeneration step requiring specific vitamin cofactors. While technically a synthetic loop, this cycle is deeply intertwined with Amino Acid Degradation, as any excess homocysteine must eventually be thoroughly degraded to prevent severe cardiovascular toxicity.
Slide 7: Amino Acid Degradation: Folic Acid and the One-Carbon Pool
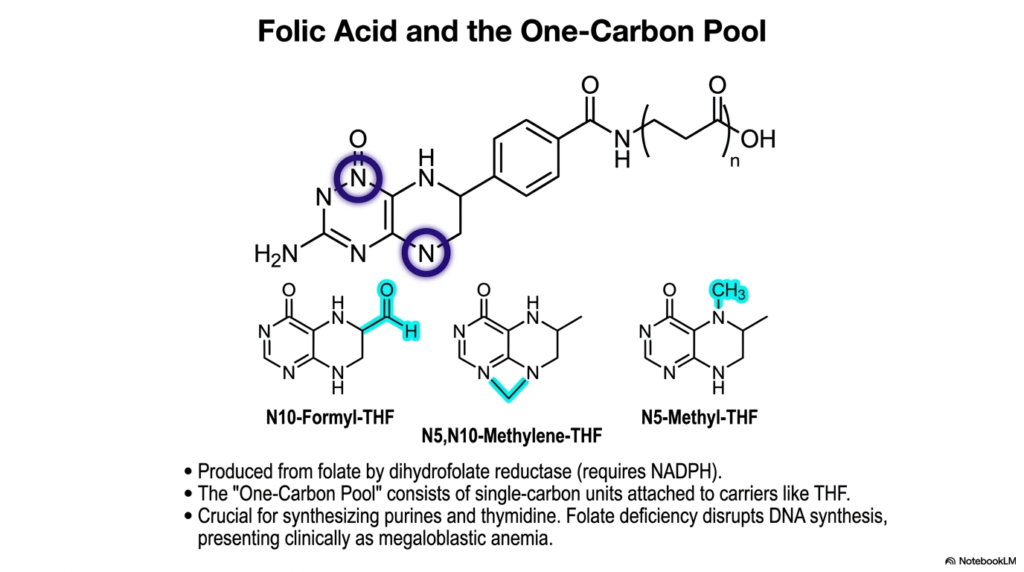
Vitamins are frequently hailed as metabolic heroes, but their true biochemical mechanisms are often misunderstood by the general public. The core purpose of this slide is to deeply illustrate the crucial enzymatic role of folic acid and the “One-Carbon Pool” in cellular metabolism, which operates closely alongside the extensive pathways of Amino Acid Degradation.
The “One-Carbon Pool” refers to a dynamic biological inventory of single-carbon units attached to specialized carrier molecules like Tetrahydrofolate (THF). These highly active carriers are enzymatically produced from dietary folate by dihydrofolate reductase, a complex reaction that strictly requires NADPH. Within the broader context of Amino Acid Degradation, THF derivatives act as molecular taxis, efficiently moving single carbons derived from amino acid catabolism to exactly where they are needed most.
These THF carriers exist in several distinct oxidation states, including N10-Formyl-THF, N5-Methylene-THF, and N5-Methyl-THF. Each specific molecular derivative has a completely distinct biochemical destiny. For example, some are absolutely crucial for the biosynthesis of purines and thymidine, meaning that rapid cellular division and healthy DNA replication heavily rely on the steady supply of these carbon units generated during metabolic breakdown.
When the delicate physiological balance between Amino Acid Degradation and one-carbon metabolism is disrupted, the clinical consequences are profoundly severe. Folate deficiency directly halts the production of these critical THF carriers, thereby disrupting DNA synthesis throughout the body. Clinically, this catastrophic metabolic failure presents as megaloblastic anemia, vividly demonstrating exactly why maintaining the one-carbon pool is absolutely essential for rapidly dividing cellular populations.
Slide 8: Amino Acid Degradation: BCAA Catabolism in Peripheral Tissues
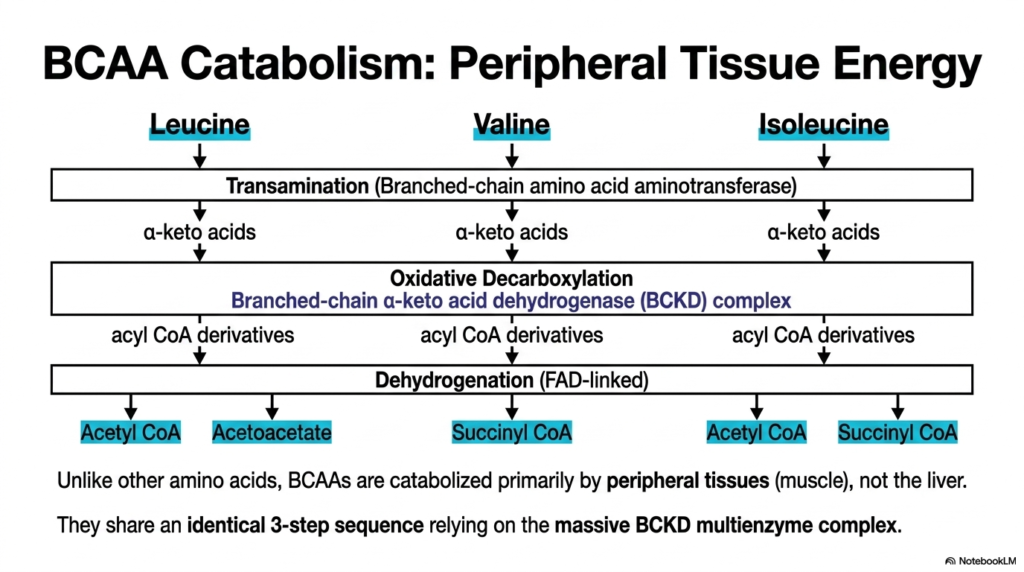
Most metabolic heavy lifting occurs deep within the liver, making it the undisputed central processing hub of human biochemistry. However, the core purpose of this slide is to highlight a major, fascinating exception to Amino Acid Degradation: the targeted catabolism of Branched-Chain Amino Acids (BCAAs), which occurs primarily in peripheral skeletal muscle.
Unlike the vast majority of other metabolic substrates, leucine, valine, and isoleucine bypass hepatic metabolism entirely. Their specific Amino Acid Degradation begins forcefully in the skeletal muscle with an initial transamination step catalyzed by branched-chain amino acid aminotransferase. This specific localized reaction heavily generates branched-chain alpha-keto acids, immediately providing working muscles with a highly accessible and rapid source of metabolic fuel during intense physiological stress.
Following transamination, these modified alpha-keto acids undergo a critical, highly regulated oxidative decarboxylation step. This irreversible biochemical reaction is carried out by the large Branched-chain alpha-keto acid dehydrogenase (BCKD) multienzyme complex. The remarkable efficiency of Amino Acid Degradation is evident here, as all three distinct BCAAs remarkably share this identical three-step enzymatic sequence, relying entirely on the BCKD complex to progress further into intermediary metabolism.
The final dehydrogenation step seamlessly produces acyl CoA derivatives that eventually yield acetyl CoA, acetoacetate, or succinyl CoA. By strictly localizing this specific branch of Amino Acid Degradation within peripheral tissues, the robust human body ensures that skeletal muscles have direct, immediate access to energy-dense carbon skeletons. This unique tissue distribution heavily highlights the incredible cellular specialization and spatial organization of human metabolic pathways.
Slide 9: Amino Acid Degradation: BCKD Deficiency and MSUD
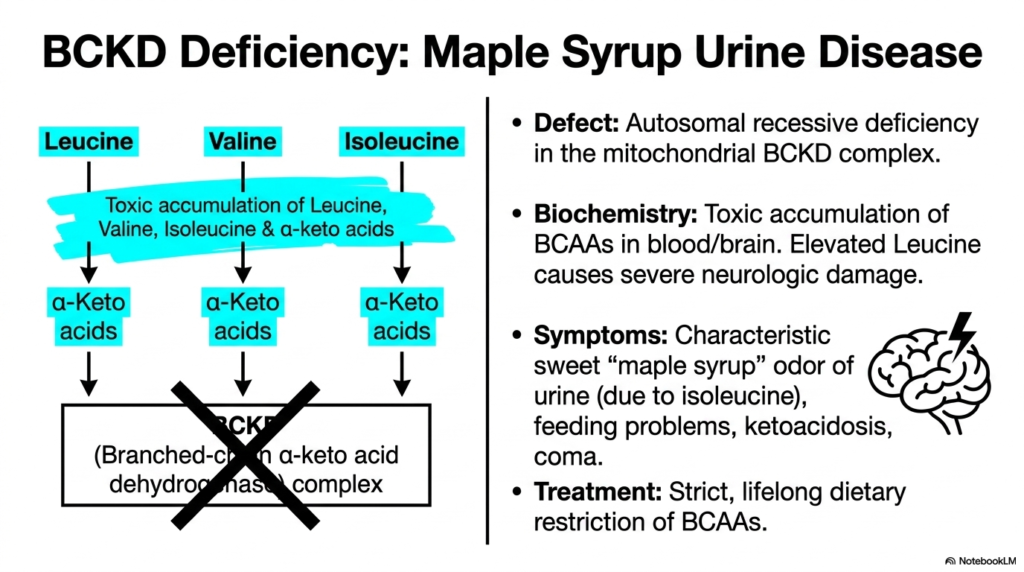
Enzymatic complexes are incredibly intricate biochemical machines, and even a single genetic mutation can bring entire metabolic pathways to a catastrophic halt. The core purpose of this slide is to carefully examine Maple Syrup Urine Disease (MSUD), a severe clinical pathology directly caused by a critical enzymatic failure in the Amino Acid Degradation of BCAAs.
Maple Syrup Urine Disease strictly arises from a devastating autosomal recessive deficiency in the mitochondrial BCKD complex. When this crucial multi-enzyme machine fails, the normal physiological progression of Amino Acid Degradation is completely and irreversibly blocked. Consequently, branched-chain amino acids, along with their highly reactive alpha-keto acid counterparts, cannot be further metabolized and rapidly accumulate in the bloodstream and central nervous system.
The biochemistry of this specific Amino Acid Degradation defect is profoundly neurotoxic. The massive, unregulated buildup of leucine and its metabolites causes severe, irreversible neurologic damage if left untreated. Clinically, this rare disease derives its name from the highly characteristic sweet, maple syrup-like odor of the pediatric patient’s urine, which is a direct byproduct of accumulating isoleucine metabolites spilling heavily into the renal system.
Patients suffering from this catastrophic failure of Amino Acid Degradation typically present with severe infant feeding problems, profound ketoacidosis, and potentially fatal comas. The only effective, life-saving medical treatment requires a strict, intensely monitored lifelong dietary restriction of leucine, isoleucine, and valine. This intense nutritional management strongly prevents toxic accumulation while providing just enough substrate for normal bodily protein synthesis, highly illustrating the delicate clinical balance of metabolic disorders.
Slide 10: Amino Acid Degradation: The PAH Reaction
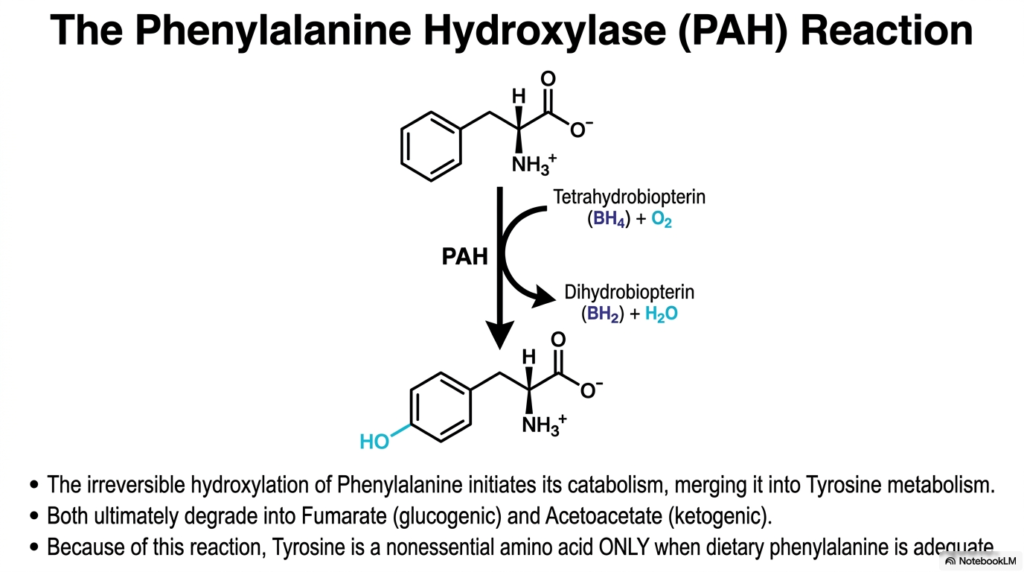
Transforming one complex organic molecule into another requires incredible enzymatic precision, especially when incorporating highly reactive oxygen atoms into aromatic rings. The core purpose of this slide is to deeply detail the Phenylalanine Hydroxylase (PAH) reaction, which serves as the strict, irreversible gateway for the targeted Amino Acid Degradation of phenylalanine into tyrosine.
The PAH enzyme smoothly facilitates the strictly irreversible hydroxylation of phenylalanine. This single, highly specific chemical reaction effectively initiates its catabolism, seamlessly merging the metabolic destiny of phenylalanine directly into the downstream tyrosine metabolism pathway. In the broader, overarching scope of Amino Acid Degradation, both of these aromatic molecular structures ultimately degrade into fumarate, which is highly glucogenic, and acetoacetate, which is purely ketogenic.
This incredibly precise hydroxylation reaction absolutely requires both molecular oxygen and a highly specialized cofactor known as Tetrahydrobiopterin (BH4). During the chemical conversion, PAH actively uses BH4 to add a hydroxyl group to the aromatic ring, thereby oxidizing the required cofactor to Dihydrobiopterin (BH2). Without this specific cofactor functioning correctly and being regenerated, the entire pathway of Amino Acid Degradation for phenylalanine grinds to an immediate and dangerous halt.
Because of this highly efficient and vital enzymatic conversion, tyrosine holds a very unique nutritional status in the human diet. Tyrosine is technically considered a nonessential amino acid strictly only when dietary phenylalanine intake is wholly adequate to physically support this PAH reaction. Understanding this specific, heavily regulated step in Amino Acid Degradation is crucial for biochemists, as it perfectly illustrates how essential nutrients can directly synthesize nonessential ones through targeted enzymatic modifications.
Slide 11: Amino Acid Degradation: PAH Deficiency and PKU
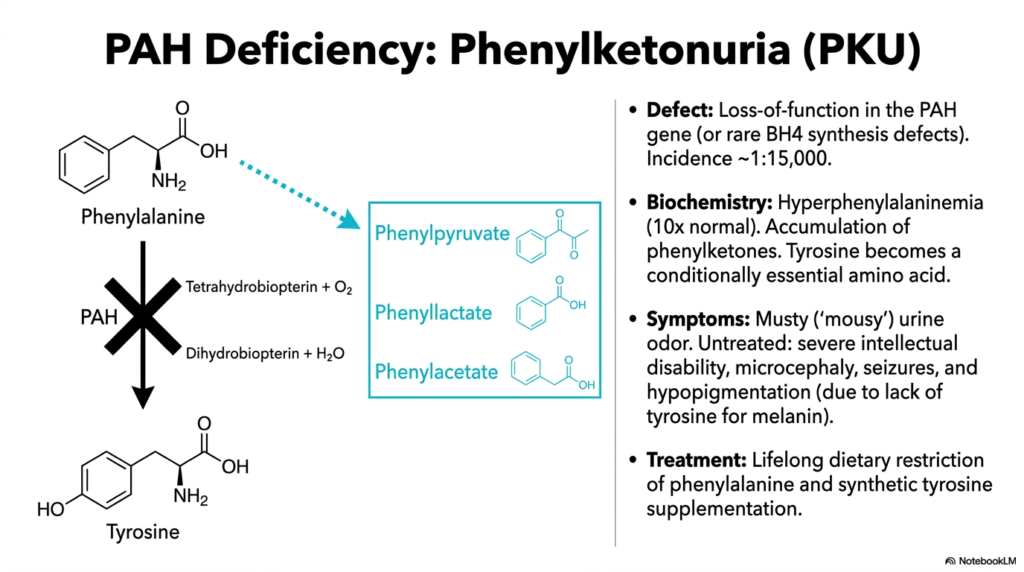
Newborn genetic screening programs have thoroughly revolutionized pediatric medicine, effectively turning potential tragedies into highly manageable metabolic conditions. The core purpose of this slide is to explore Phenylketonuria (PKU), the most widely recognized inborn error of Amino Acid Metabolism, which results directly from a devastating genetic defect in the Phenylalanine Hydroxylase (PAH) enzyme.
PKU primarily arises from a dangerous autosomal recessive loss-of-function mutation heavily impacting the PAH gene itself, though rare severe defects in BH4 cofactor synthesis can also be responsible. When this critical, gatekeeping step in Amino Acid Degradation completely fails, phenylalanine absolutely cannot be converted into tyrosine. This enzymatic blockade forcibly directs alternative, far less efficient metabolic pathways to take over, leading to severe hyperphenylalaninemia where blood levels reach ten times the normal healthy concentration.
The systemic disruption of normal Amino Acid Degradation leads to the toxic accumulation of phenylketones, such as phenylpyruvate and phenyllactate, which are massively excreted in the urine and impart a characteristic, strongly musty or ‘mousy’ odor. Furthermore, because the primary biochemical conversion pathway is entirely broken, tyrosine suddenly and dangerously becomes a conditionally essential amino acid for these patients, requiring direct, heavy dietary supplementation to support crucial neurotransmitter and melanin pigment synthesis.
If this severe genetic defect in Amino Acid Degradation is left untreated, the toxic accumulation directly causes severe intellectual disability, microcephaly, and persistent, dangerous seizures. Additionally, the sheer lack of downstream tyrosine inevitably results in systemic hypopigmentation. The absolute cornerstone of medical treatment remains the heavily monitored lifelong dietary restriction of phenylalanine combined with synthetic tyrosine supplementation, heavily preventing profound neurological damage and ensuring relatively normal cognitive development.
Slide 12: Amino Acid Degradation: Downstream Tyrosine Defects
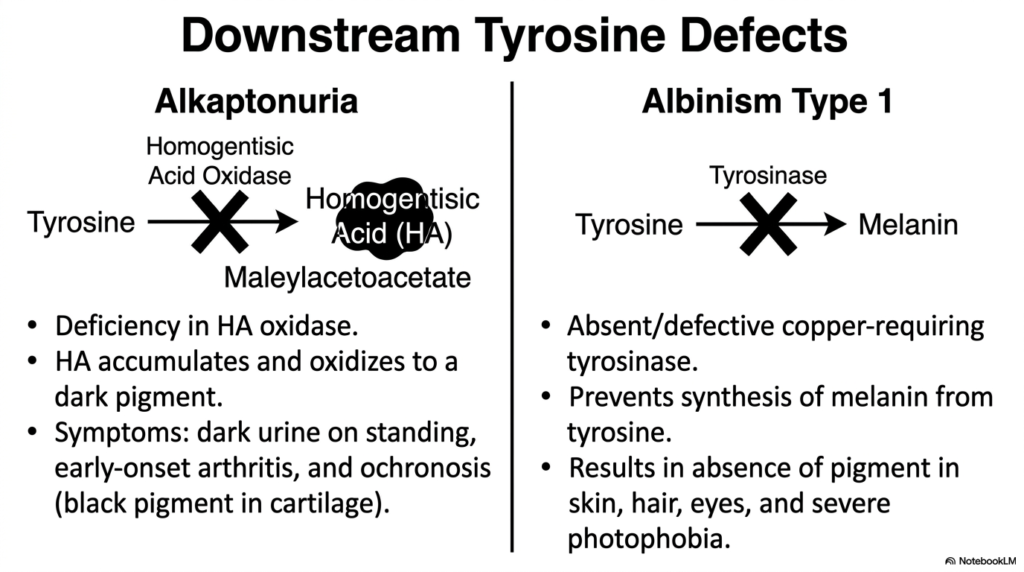
Metabolic pathways are akin to complex industrial assembly lines; a mechanical breakdown at any point can yield markedly different yet equally profound physiological consequences. The core purpose of this slide is to closely examine how distinct genetic blockades situated further down the Amino Acid Degradation pathway of tyrosine precisely result in two entirely unique clinical conditions: Alkaptonuria and Albinism.
Alkaptonuria is clinically caused by a specific genetic deficiency that severely impairs the enzyme homogentisic acid oxidase. Because this targeted step of Amino Acid Degradation is permanently blocked, homogentisic acid heavily and dangerously accumulates in tissues and oxidizes into a dark, highly reactive molecular pigment. Clinically, affected patients present with urine that turns strikingly dark upon standing, early-onset debilitating arthritis, and profound ochronosis, which is the visible, painful accumulation of black pigment in connective joint cartilages.
Conversely, Albinism Type 1 vividly represents an entirely different systemic failure strictly related to tyrosine metabolism, though branching distinctly away from core energy-yielding Amino Acid Degradation. This condition is directly caused by the absence or functional deficiency of a copper-requiring enzyme known as tyrosinase. Without this crucial, highly targeted enzymatic activity, the human body is completely and permanently unable to synthesize the dark pigment melanin from its available tyrosine precursors.
While Alkaptonuria vividly represents a severe failure of energetic Amino Acid Degradation, Albinism strongly represents a massive failure of targeted biosynthetic diversion. The absolute lack of tyrosinase strictly results in a profound, noticeable absence of pigmentation in the skin, hair, and eyes, which heavily occurs alongside severe photophobia. Comparing these two downstream defects perfectly highlights how the metabolic fate of a single molecule can drastically alter human physiology depending entirely on which specific enzymatic pathway fails.
Slide 13: Amino Acid Degradation: Cystathionine Beta-Synthase Deficiency
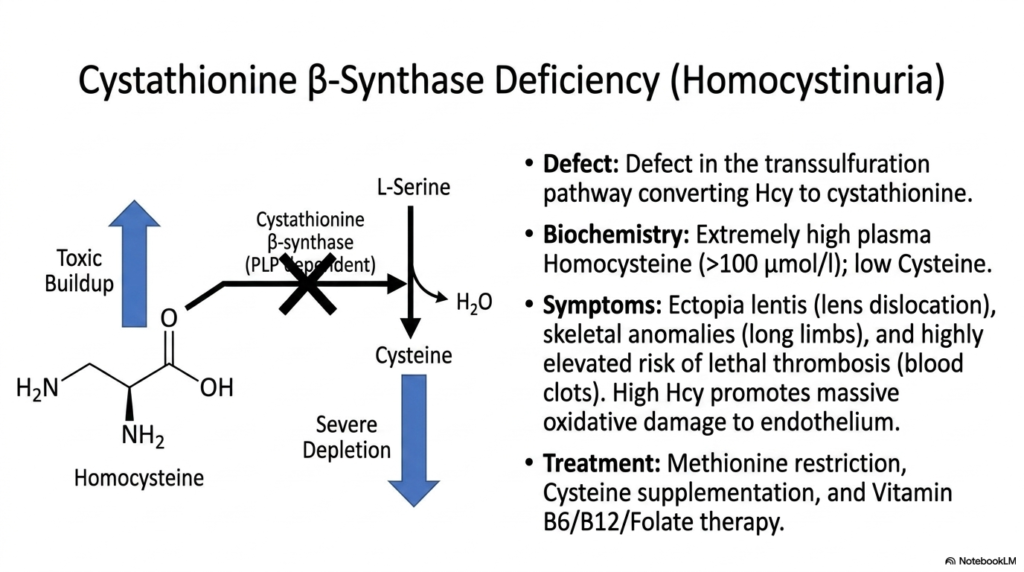
Cardiovascular health is deeply and inextricably intertwined with microscopic metabolic processes that occur continuously within delicate cellular tissues. The core purpose of this slide is to deeply explore Homocystinuria, a severe, life-threatening clinical disorder primarily resulting from a critical genetic blockade in the transsulfuration pathway, a vital sub-route of broader Amino Acid Degradation.
Homocystinuria is primarily and heavily caused by a severe genetic defect in cystathionine beta-synthase, a strictly PLP-dependent enzyme. In healthy, normal Amino Acid Degradation, this highly active enzyme safely and efficiently converts toxic homocysteine into harmless cystathionine by condensing it intimately with serine. When this vital enzyme utterly fails, the entire transsulfuration pathway completely collapses, leading to severely depleted downstream cysteine levels and an extremely dangerous, massive vascular buildup of plasma homocysteine.
The biochemical consequences of this dangerously disrupted Amino Acid Degradation are incredibly destructive to the human body. High circulating homocysteine levels aggressively promote extensive oxidative damage, heavily targeting the delicate vascular endothelium. Clinically, this presents an extraordinarily high risk of premature, highly lethal thrombosis and systemic blood clots. Patients vividly exhibit distinct physical anomalies as well, including ectopia lentis (lens dislocation) and significantly elongated skeletal limbs closely resembling Marfan syndrome.
Treating this exceptionally dangerous disruption in Amino Acid Degradation absolutely requires intensive, lifelong metabolic management. Physicians typically strictly prescribe heavy methionine restriction to immediately limit upstream precursors, alongside mandatory cysteine supplementation to artificially replace the missing downstream product. Additionally, extremely high-dose Vitamin B6, B12, and Folate therapies are frequently administered to forcefully stimulate any residual enzymatic activity and push alternative remethylation pathways, heavily protecting the patient’s highly vulnerable vascular system.
Slide 14: Amino Acid Degradation: Biosynthesis from Intermediates
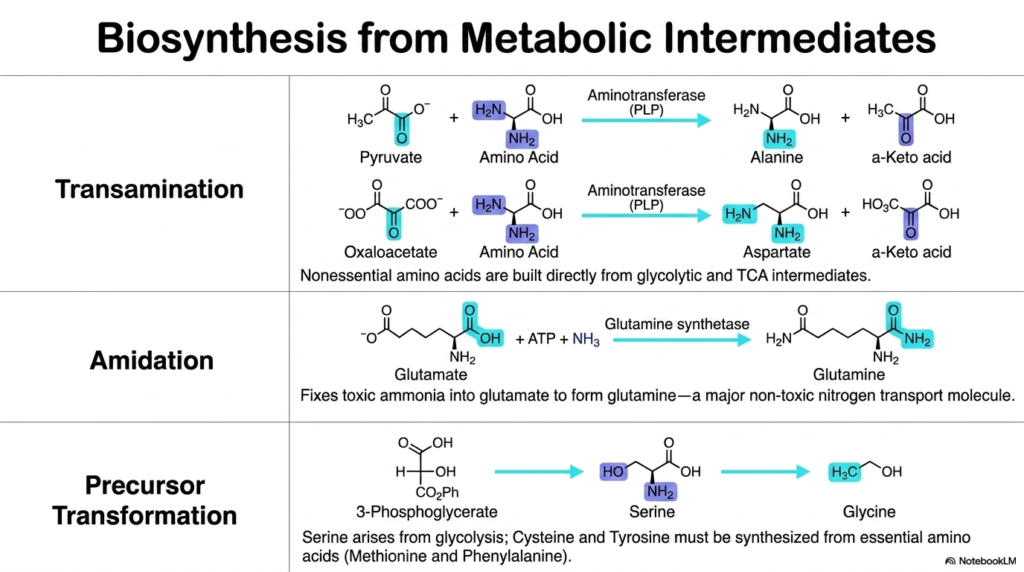
Metabolism is fundamentally and deeply a two-way street, constantly and heavily balancing the extensive breakdown of complex molecules with the continuous, vital synthesis of new ones. The core purpose of this slide is to sharply pivot from pure Amino Acid Degradation to heavily demonstrate exactly how the human body ingeniously synthesizes entirely nonessential amino acids directly from highly common metabolic intermediates.
The elegant, highly efficient symmetry of cellular metabolism strongly dictates that many core pathways of Amino Acid Degradation can run smoothly and efficiently in reverse. Through the highly regulated process of transamination, crucial nonessential amino acids are built directly from highly accessible glycolytic and TCA intermediates. For example, common molecules like pyruvate and oxaloacetate can quickly accept an amino group via PLP-dependent aminotransferases, instantly generating alanine and aspartate, respectively, to rapidly meet the body’s immediate systemic biosynthetic demands.
Another critically important synthetic mechanism closely linked to Amino Acid Degradation is amidation. Through the targeted action of glutamine synthetase, highly toxic free ammonia is actively fixed securely onto glutamate to form glutamine. This ATP-dependent reaction not only synthesizes a crucially needed amino acid but also brilliantly creates a major, completely non-toxic nitrogen transport molecule that safely ferries volatile ammonia through the bloodstream without ever damaging delicate neurological tissues.
Finally, complex precursor transformation strongly showcases the incredible adaptability of human biochemistry. Serine is derived smoothly and efficiently from the common glycolytic intermediate 3-phosphoglycerate. However, highly complex molecules like cysteine and tyrosine absolutely must be carefully synthesized from essential amino acids like methionine and phenylalanine, respectively. This intricate biological web strongly ensures that the valuable byproducts of Amino Acid Degradation are continuously and effectively recycled, maintaining a highly robust supply of building blocks for vital cellular structures.
Slide 15: Amino Acid Degradation: Inborn Errors Summary
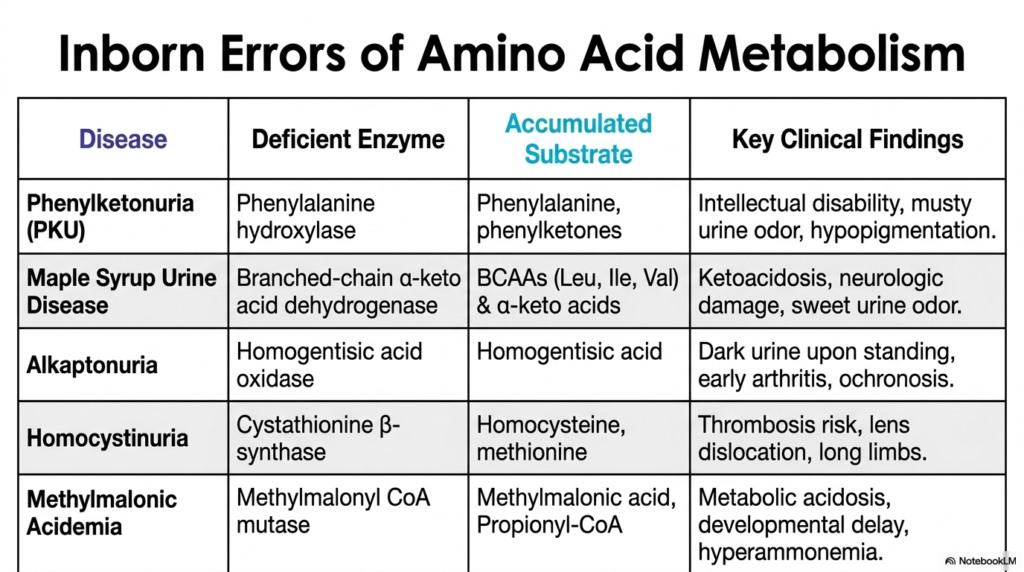
Mastering clinical biochemistry requires an innate ability to quickly synthesize incredibly complex metabolic pathways into actionable, diagnostic knowledge. The core purpose of this slide is to effectively provide a highly comprehensive, high-yield summary of the major inborn errors of Amino Acid Degradation. This strictly serves as a vital review tool for dedicated medical and biochemistry students preparing for rigorous clinical board examinations.
The catastrophic, systemic failure of Amino Acid Degradation almost invariably leads to highly severe clinical phenotypes directly due to the incredibly toxic accumulation of highly specific substrates. Phenylketonuria (PKU), caused by deficient phenylalanine hydroxylase, results in the dangerous buildup of phenylalanine, presenting clinically with severe intellectual disability and pronounced hypopigmentation. Similarly, Maple Syrup Urine Disease rapidly halts branched-chain alpha-keto acid dehydrogenase, strongly allowing neurotoxic BCAAs to aggressively trigger highly fatal ketoacidosis and brain damage.
Other specific genetic defects that heavily impact Amino Acid Degradation present with highly distinctive systemic symptoms. Alkaptonuria, resulting from a total loss of homogentisic acid oxidase, causes dark urine and severely debilitating early-onset arthritis, specifically due to heavy pigment accumulation. Homocystinuria, heavily caused by severely deficient cystathionine beta-synthase, aggressively drives massive homocysteine accumulation, firmly putting patients at an extreme, highly lethal risk for vascular thrombosis and distinct skeletal abnormalities like abnormally long limbs.
Finally, Methylmalonic Acidemia strongly highlights a rare genetic defect located much further down the catabolic chain, strictly involving methylmalonyl CoA mutase. This highly specific, devastating failure tightly embedded in Amino Acid Degradation firmly leads to extremely severe metabolic acidosis and life-threatening hyperammonemia. By thoroughly and strictly reviewing this consolidated table, future pediatric physicians can instantly link highly specific deficient enzymes and dangerously accumulated substrates directly to their characteristic clinical findings, strongly solidifying their mastery of these complex and life-threatening metabolic diseases.
Please read our Content Disclaimer Statement.
Check out our social media channels: