25. Enzyme Catalysis: Mechanisms and Active Site Kinetics
Imagine a bustling metropolis where millions of crucial deliveries happen every microsecond. Without a highly coordinated system, the city would collapse instantly. The human body operates similarly, relying on biological catalysts to accelerate vital chemical reactions that would otherwise take millennia. This comprehensive slide deck explores the molecular mechanics of these essential proteins. Designed for college and medical students, this guide breaks down active site kinetics, thermodynamic principles, and complex catalytic mechanisms. Let us dissect the intricate molecular machinery that effectively sustains all human life.
Slide 1: Introduction to Enzyme Catalysis Mechanics
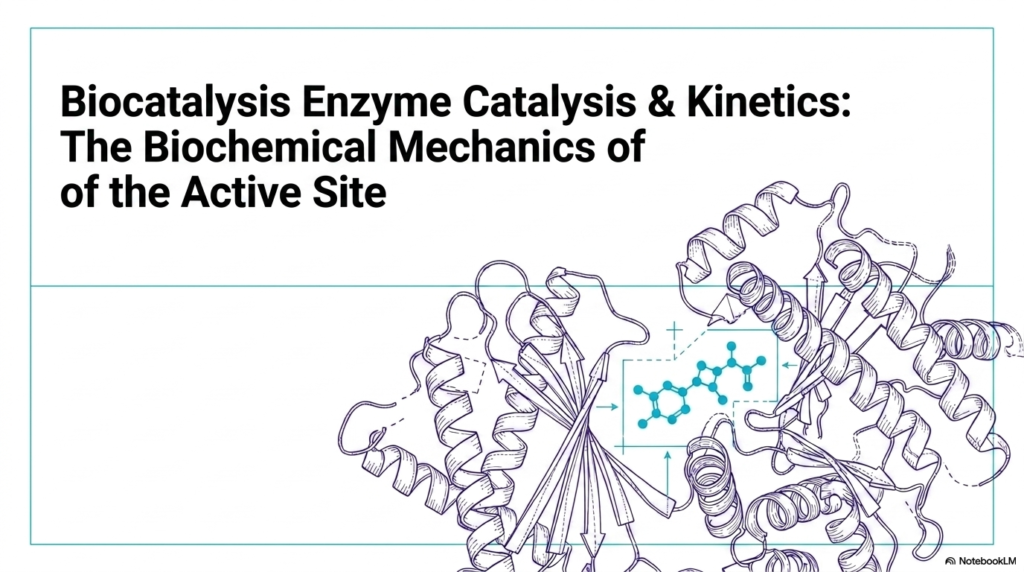
Welcome to the fascinating realm of biological acceleration. The opening slide introduces the overarching theme of Enzyme Catalysis, presenting a visually striking representation of a folded protein structure. For medical and college students, understanding this architectural complexity is the first step toward mastering biochemistry. The intricate loops and helices form a highly specialized microenvironment known as the active site. This specific three-dimensional arrangement dictates how biological molecules interact, ensuring that life-sustaining reactions occur at the exact right moment.
Without efficient Enzyme Catalysis, the fundamental pathways of metabolism and cellular respiration would grind to a halt. The study of Enzyme Catalysis goes far beyond simple lock-and-key memorization. It requires a deep dive into the kinetic principles that govern how quickly and efficiently a biological catalyst operates. As depicted in the slide’s complex molecular model, the active site is not merely a static pocket. It is a highly dynamic arena where substrate molecules are bound, manipulated, and transformed.
The protein’s structural scaffolding positions specific amino acid side chains in precise orientations, setting the stage for remarkable chemical transformations. Grasping these structural concepts is absolutely crucial for future healthcare professionals and researchers. When students analyze the visual representation on this slide, they are looking at the molecular targets for countless pharmaceutical drugs. Many life-saving medications function by interacting directly with these protein structures, either enhancing or inhibiting their natural activity.
By establishing a strong foundational understanding of how these molecules fold and function, learners can better appreciate the sophisticated biochemical mechanics that drive human physiology and disease states. Furthermore, the illustration highlights the immense difference in scale between the large protein scaffolding and the small chemical substrate it acts upon. This disparity reveals a critical concept: the vast majority of the protein’s mass is dedicated entirely to maintaining the perfect geometry of the active site.
Slide 2: The Thermodynamic Core of Enzyme Catalysis
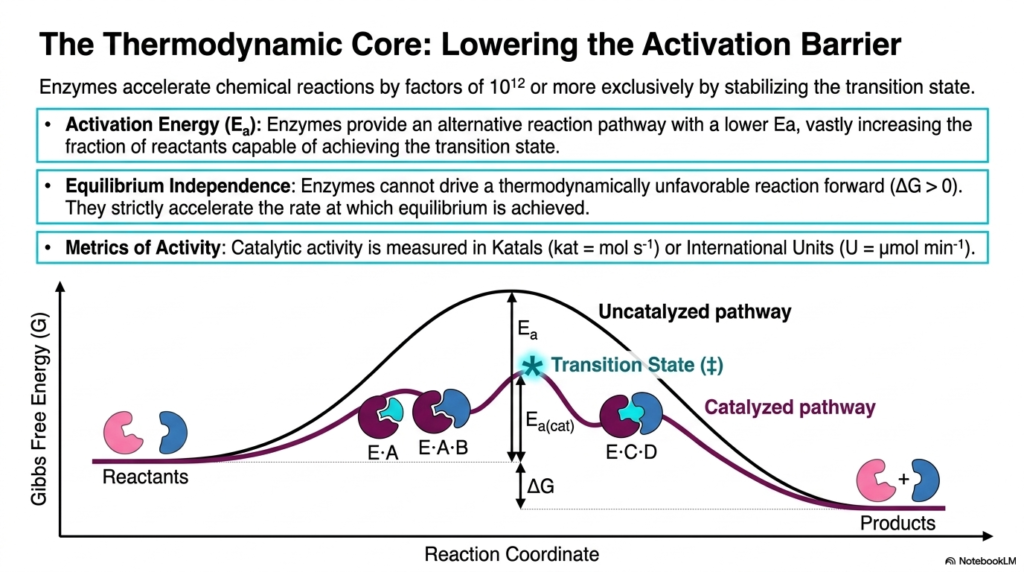
The fundamental principle of Enzyme Catalysis revolves around the manipulation of energy barriers. This slide illustrates the thermodynamic core of the process, highlighting how biological catalysts accelerate chemical reactions by factors of a trillion or more. The central mechanism is the lowering of activation energy, denoted as Ea. In the provided graph, the uncatalyzed pathway resembles a steep mountain, requiring a substantial input of energy for the reactants to reach the transition state. The catalyzed pathway offers an alternative, much lower route, vastly increasing the fraction of molecules that react.
A common misconception among biochemistry students is that these proteins alter the overall energy yield of a reaction. This slide definitively corrects that notion by illustrating equilibrium independence in Enzyme Catalysis. The Gibbs free energy difference between the reactants and products remains completely unchanged. These biological catalysts cannot force a thermodynamically unfavorable reaction to move forward; they strictly accelerate the rate at which the system reaches equilibrium. Both pathways start and end at the same energy level, differing only in the transition-state barrier.
To quantify this incredible acceleration, biochemists utilize specific metrics to measure the efficiency of Enzyme Catalysis. The slide introduces the Katal, defined as moles per second, and the International Unit, defined as micromoles per minute. These standardized units allow researchers and clinicians to measure activity accurately. By stabilizing the high-energy, unstable transition state, the biological catalyst ensures that reactants are swiftly converted into essential products. This rapid turnover is what sustains the high-speed metabolic demands of living organisms.
Understanding the energy landscape is vital for grasping how physiological systems operate under standard conditions. Without this stabilization of the transition state, the body would need dangerously high temperatures to drive metabolic reactions, ultimately denaturing proteins and destroying cells. Thus, the ability to lower the activation barrier without altering overall thermodynamics is a true cornerstone concept in modern medical science and structural biochemistry.
Slide 3: The EC Taxonomic System in Enzyme Catalysis
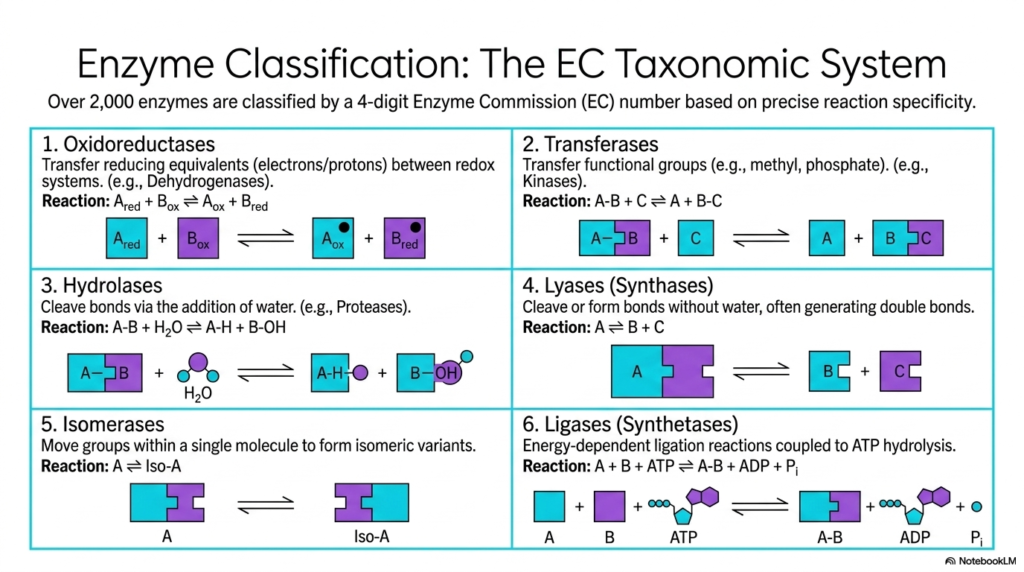
Given the sheer diversity of metabolic reactions, scientists require a rigorous classification system to organize the over 2,000 known biological catalysts. This slide introduces the Enzyme Commission taxonomic system, a fundamental framework in the study of Enzyme Catalysis. This four-digit numbering system categorizes proteins based on their precise reaction specificity. The first broad category, Oxidoreductases, involves the transfer of reducing equivalents, such as electrons or protons, between redox systems. Dehydrogenases are classic examples, playing critical roles in cellular respiration by shifting electrons to energy carriers.
The second and third categories encompass Transferases and Hydrolases. Transferases are responsible for moving functional groups, such as methyl or phosphate groups, from one molecule to another. Kinases, which transfer phosphate groups from ATP to target molecules, act as master regulators in cellular signaling. Hydrolases utilize water to cleave chemical bonds. Proteases, which break down dietary proteins in the digestive tract, utilize this specific form of Enzyme Catalysis to ensure nutrients are properly broken down and safely absorbed by the intestines.
The remaining classifications highlight even more specialized forms of Enzyme Catalysis. Lyases, or synthases, cleave or form bonds without the addition of water, often generating double bonds in the process. Isomerases perform the fascinating task of moving functional groups within a single molecule to create isomeric variants, essentially rearranging the chemical furniture without adding or removing atoms. Finally, Ligases, or synthetases, handle energy-dependent ligation reactions. These proteins couple the joining of two molecules with the hydrolysis of ATP.
Mastering this six-part classification system provides medical students with a universal language for biochemistry. Whether analyzing a standard metabolic panel or researching a novel genetic disorder, knowing which class a biological catalyst belongs to immediately reveals its fundamental mechanism of action and its role within the larger cellular ecosystem. This knowledge enables practitioners to trace metabolic pathways with total structural confidence.
Slide 4: Substrate Recognition in Enzyme Catalysis
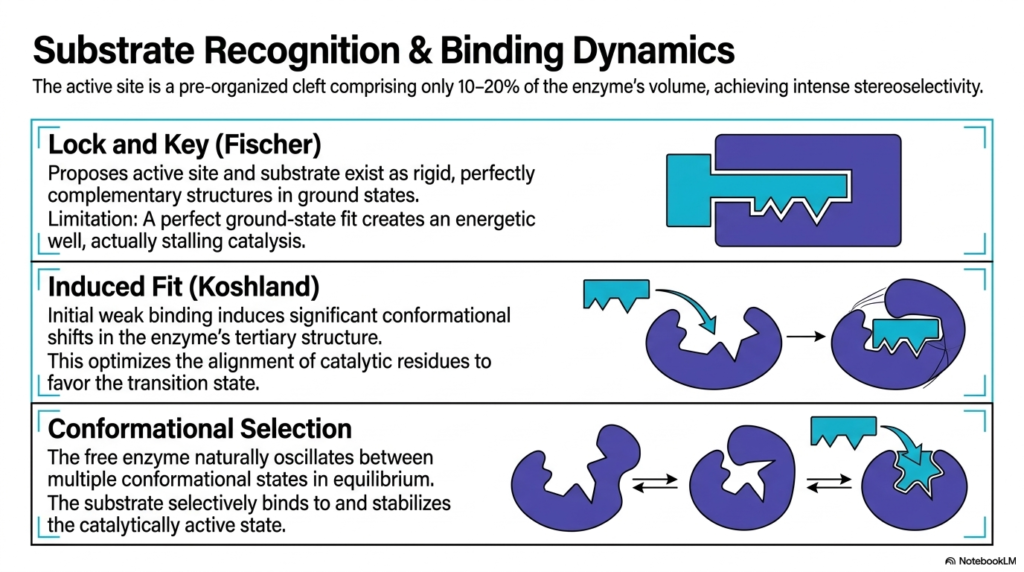
Substrate recognition is a highly specialized process, and this slide illustrates the evolutionary refinement of models explaining Enzyme Catalysis. The active site is a preorganized cleft that occupies only a fraction of the protein’s total volume, yet it exhibits high stereoselectivity. Historically, the Lock and Key model proposed by Fischer suggested that the active site and the substrate existed as rigid, perfectly complementary structures. While intuitive, this early model had a fatal limitation: a perfect ground-state fit creates an energetic well, actually stalling the reaction completely.
To resolve this thermodynamic trap, Koshland introduced the Induced Fit model, which drastically shifted our understanding of Enzyme Catalysis. This modern paradigm proposes that initial, weak binding between the molecule and the protein induces significant conformational shifts in the enzyme’s tertiary structure. The active site essentially wraps around the substrate, molding itself to achieve the perfect functional fit. This dynamic shifting optimizes the alignment of catalytic residues, pushing the energetic favorability away from the ground state and strongly toward the highly reactive transition state.
The slide also details the most contemporary framework, Conformational Selection. This sophisticated view of Enzyme Catalysis acknowledges that a free protein naturally oscillates between multiple conformational states in a dynamic equilibrium. The substrate does not force a shape change from a completely static state; rather, it selectively binds to and stabilizes the specific transient conformation that happens to be catalytically active at that millisecond. This framework highlights the incredible flexibility and dynamic nature of folded protein structures in aqueous cellular environments.
For college students, recognizing the shift from rigid models to dynamic conformational frameworks is essential. Proteins are not static puzzle pieces; they are highly fluid molecular machines that breathe, flex, and constantly adapt. This dynamic behavior explains how biological catalysts can achieve such astonishing specificity and speed, differentiating between nearly identical molecules with flawless chemical precision.
Slide 5: Non-Covalent Orchestration in Enzyme Catalysis
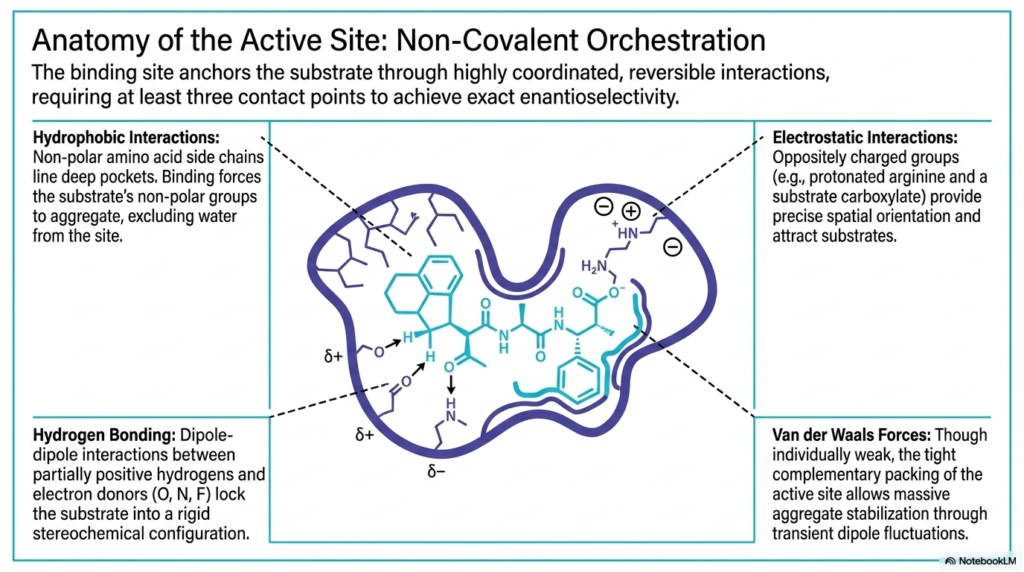
The anatomy of the active site is a masterpiece of molecular architecture. This slide delves into the precise chemical forces that anchor the substrate during Enzyme Catalysis. The binding site requires highly coordinated, reversible interactions, utilizing at least three distinct contact points to achieve exact enantioselectivity. Hydrophobic interactions play a massive role here. Non-polar amino acid side chains line deep pockets within the protein, forcing the substrate’s non-polar groups to aggregate. This strategic arrangement effectively excludes water from the active site, creating an isolated microenvironment.
Hydrogen bonding provides another crucial layer of structural control during Enzyme Catalysis. These dipole-dipole interactions occur between partially positive hydrogens and electron donors like oxygen, nitrogen, or fluorine. By forming these specific directional bonds, the active site locks the substrate into a rigid stereochemical configuration. Electrostatic interactions further enhance this precision. Oppositely charged groups, such as a protonated arginine on the protein and a carboxylate on the substrate, attract each other with significant force, providing exact spatial orientation as the molecule settles into the cleft.
Finally, the slide highlights the subtle but powerful role of Van der Waals forces in Enzyme Catalysis. While individually weak and transient, these forces become incredibly significant due to the tight, complementary packing of the active site. When the substrate is perfectly nestled within the protein cleft, the sheer surface area of contact enables robust aggregate stabilization via transient dipole fluctuations. This combination of weak, reversible forces ensures that the substrate is held tightly enough to react but loosely enough for products to eventually be released.
Understanding this non-covalent symphony is fundamental for modern pharmacology. Drug designers rely heavily on these exact same interactions when synthesizing competitive inhibitors. By mapping the hydrophobic pockets, hydrogen-bond donors, and electrostatic charges of an active site, scientists can engineer targeted pharmaceutical compounds that bind with even greater affinity than the natural biological substrate.
Slide 6: The Transition State Paradox in Enzyme Catalysis
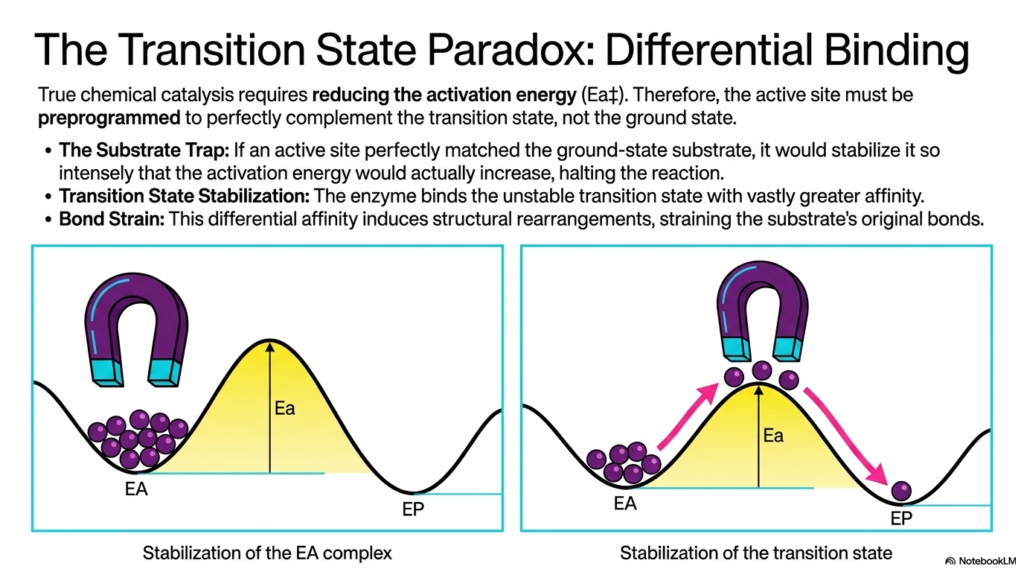
True chemical acceleration requires reducing the activation energy, leading to a fascinating thermodynamic challenge known as the transition state paradox. This slide explains a foundational rule of Enzyme Catalysis: the active site must be preprogrammed to perfectly complement the transition state, not the ground state. If the active site were a perfect match for the initial substrate, it would create a thermodynamic “substrate trap.” The intense stabilization of the ground state would actually increase the activation energy required to proceed, effectively halting the chemical reaction in its tracks.
To avoid this energetic trap, Enzyme Catalysis relies on differential binding affinity. The biological catalyst is structured to bind the highly unstable transition state with vastly greater affinity than it binds the initial reactants. The visual analogy of the magnet beautifully illustrates this core concept. By pulling the reacting molecules toward a high-energy geometry, the active site essentially drags the substrate up the energetic hill. This selective stabilization significantly lowers the activation peak, allowing a much larger percentage of molecules to successfully cross the threshold.
This differential affinity inherently induces structural rearrangements, resulting in significant bond strain. As the active site forces the substrate into the transition state geometry, the original chemical bonds of the reactant are intensely stretched, twisted, and weakened. This mechanical and electrostatic stress is a primary driver of Enzyme Catalysis. By destabilizing the starting material and heavily rewarding the formation of the transition state, the protein expertly manipulates the energetic landscape to favor incredibly rapid chemical turnover.
This paradox is a brilliant example of evolutionary engineering. For medical students, mastering this specific concept helps explain why transition-state analogs are the most potent pharmaceutical inhibitors. By synthesizing a synthetic drug that mimics the geometry of the transition state rather than the substrate, researchers can create medications that bind to the active site almost irreversibly.
Slide 7: Proximity and Desolvation Mechanisms in Enzyme Catalysis
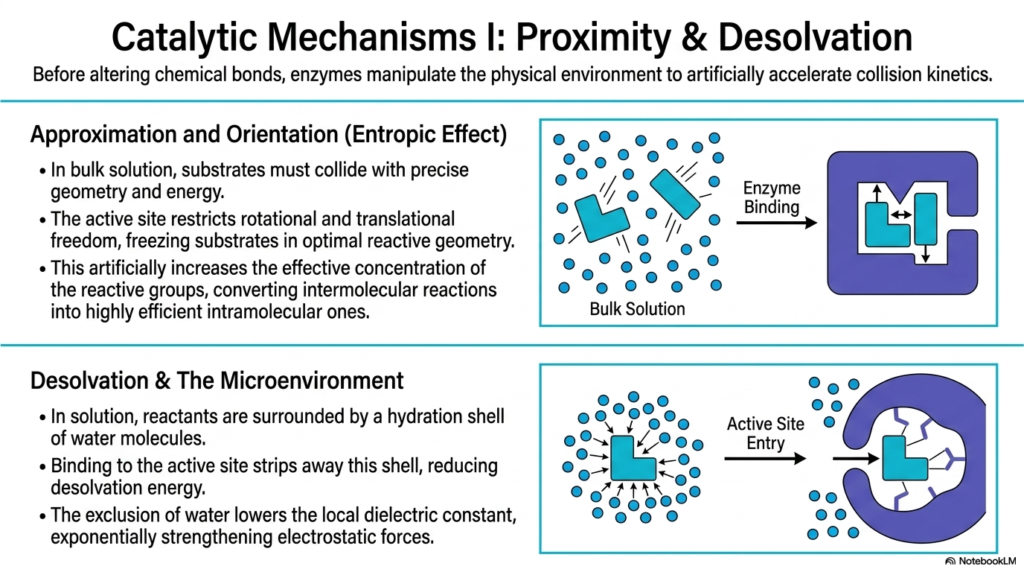
Before directly altering chemical bonds, biological catalysts powerfully manipulate the physical environment to artificially accelerate collision kinetics. This slide details the first major set of mechanisms in Enzyme Catalysis: proximity and desolvation. In a bulk aqueous solution, substrates must collide randomly, in the correct geometry, and with sufficient kinetic energy to react. The active site overcomes this massive entropic hurdle through approximation and orientation. By binding the substrates, the protein restricts their rotational and translational freedom, freezing them in the optimal reactive geometry.
This spatial restriction artificially increases the effective concentration of the reactive groups. What would normally be a slow, random intermolecular reaction in the cellular fluid is brilliantly converted into a highly efficient, fast intramolecular reaction within the active site. This entropic effect is a massive contributor to the sheer speed of Enzyme Catalysis. By completely removing the randomness of molecular motion, the biological catalyst ensures that when two substrates are held together, they are perfectly aligned for an immediate chemical transformation.
Furthermore, the slide explores the critical process of desolvation and the creation of a specialized microenvironment. In a standard cellular solution, reactants are encased in a dense hydration shell of water molecules. Binding to the active site forcefully strips away this protective shell, reducing the desolvation energy required for the reaction to proceed. This absolute exclusion of water drastically lowers the local dielectric constant within the cleft, a true hallmark feature of successful Enzyme Catalysis.
Lowering the local dielectric constant by removing water exponentially strengthens the electrostatic forces between the substrate and the catalytic amino acid residues. In this highly specialized anhydrous microenvironment, electrical charges are no longer masked by surrounding water dipoles. This allows for incredibly intense and precise chemical interactions between the protein and the substrate, perfectly setting the stage for the direct bond-breaking mechanisms that follow.
Slide 8: Acid-Base and Covalent Strategies in Enzyme Catalysis
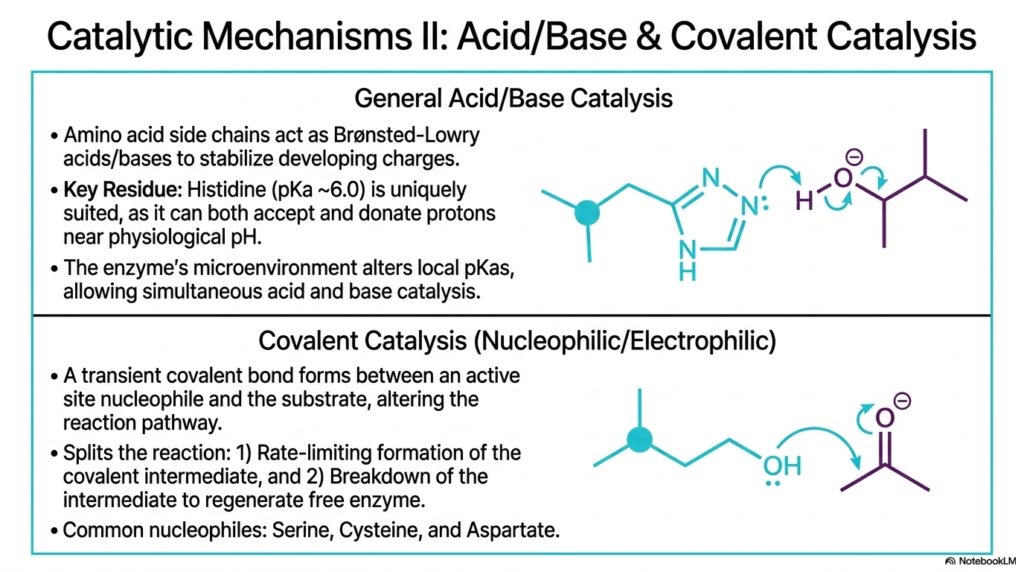
Moving beyond spatial manipulation, this slide transitions to the direct chemical mechanisms involved in Enzyme Catalysis. The first major strategy discussed is general acid-base catalysis. In this process, specific amino acid side chains within the active site act as Brønsted-Lowry acids or bases, swiftly donating or accepting protons to stabilize developing electrical charges during the reaction. Histidine is a uniquely suited key residue for this task. With a pKa near 6.0, it can act as both a proton donor and an acceptor at physiological pH.
The unique microenvironment of the active site plays a major role by altering the local pKa values of surrounding amino acid side chains. This allows the protein to conduct simultaneous acid-base chemistry, a feat that is nearly impossible in a standard bulk solution. The second major strategy driving Enzyme Catalysis is covalent, or nucleophilic, catalysis. This involves the formation of a transient covalent bond between a highly reactive nucleophile at a reactive site and the substrate molecule, fundamentally altering the reaction pathway.
Covalent Enzyme Catalysis essentially splits a difficult, high-energy single-step reaction into a faster, two-step process. First, there is the rate-limiting formation of the temporary covalent intermediate between the protein and the substrate. Second, the rapid breakdown of this intermediate yields the final biochemical product and safely regenerates the free, active biological catalyst. Common nucleophiles used by proteins in this mechanism include the hydroxyl group of Serine, the thiol group of Cysteine, and the carboxylate group of Aspartate.
For medical students, a deep understanding of these specific chemical strategies is vital to grasping advanced enzyme kinetics and pharmacology. Many potent irreversible inhibitors and cellular toxins work by specifically attacking these highly reactive nucleophilic residues, permanently capping them with a rigid covalent bond. Recognizing these vulnerable active site residues helps in anticipating potential drug interactions and understanding severe toxicological vulnerabilities in patients.
Slide 9: Electrostatics and Metal Ions in Enzyme Catalysis
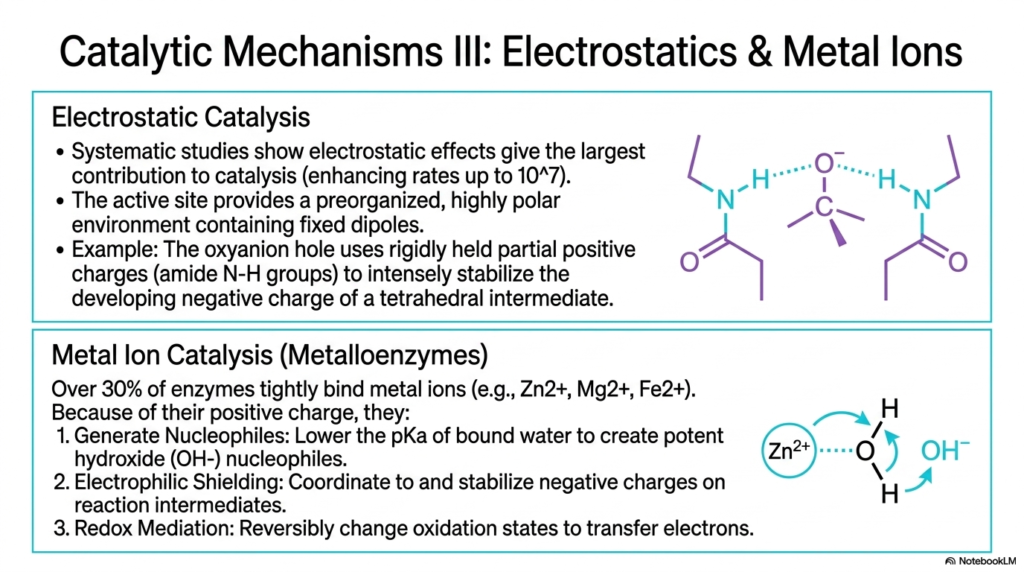
Systematic studies have revealed that electrostatic effects often provide the absolute largest single contribution to reaction acceleration. This slide delves into the third major mechanism of Enzyme Catalysis, focusing on electrostatics and the vital role of metal ions. The active site provides a pre-organized, highly polar environment filled with fixed dipoles. A classic example is the oxyanion hole, an architectural feature that uses rigidly held partial positive charges, typically from amide nitrogen-hydrogen groups, to intensely stabilize the developing negative charge of a tetrahedral transition state.
By neutralizing these explosive, high-energy charges during the transition state, electrostatic Enzyme Catalysis prevents the reaction intermediate from breaking down prematurely. The second portion of the slide introduces metal ion catalysis, characteristic of metalloenzymes. Over 30% of known biological catalysts tightly bind trace metal ions such as zinc, magnesium, and iron. Because of their concentrated positive charge, these inorganic elements provide exceptional chemical versatility that standard amino acid side chains simply cannot replicate in biological systems.
These tightly bound metal ions accelerate Enzyme Catalysis through several distinct, powerful pathways. First, they can generate potent nucleophiles; for example, binding water to a zinc ion drastically lowers its pKa, creating a highly reactive hydroxide ion ready for immediate attack. Second, metals provide strong electrophilic shielding, coordinating to and stabilizing negative charges on reaction intermediates far better than amino acids alone can. Finally, transition metals act as excellent redox mediators, reversibly changing their oxidation states to rapidly shuttle electrons.
This incredible reliance on metal ions underscores the biological necessity of dietary trace minerals. A severe deficiency in zinc or magnesium can profoundly impair the function of a vast array of metalloenzymes, leading to widespread cellular and metabolic dysfunction. Understanding this relationship beautifully bridges the gap between molecular biochemistry and clinical nutrition for aspiring medical professionals, proving how fundamental diet is to cellular machinery.
Slide 10: Cofactors and Coenzymes in Enzyme Catalysis
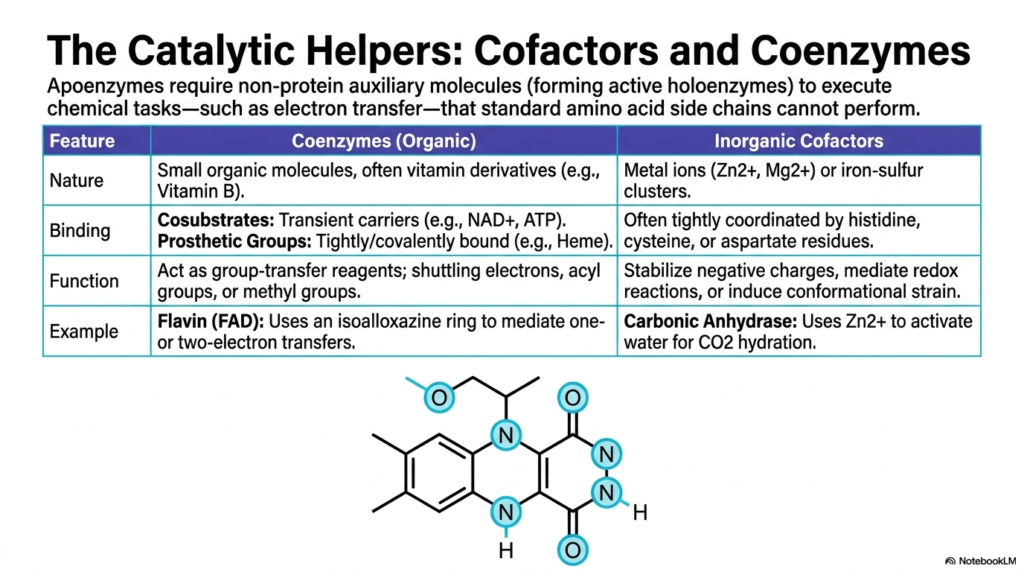
Sometimes, the standard twenty amino acids are completely insufficient to execute complex chemical tasks, particularly those involving intricate electron transfer. This slide introduces the catalytic helpers essential to enzyme catalysis: cofactors and coenzymes. Without these non-protein auxiliary molecules, many cellular proteins exist as inactive apoenzymes. When the necessary helper molecule binds, the complex becomes a fully active holoenzyme, capable of driving life-sustaining reactions. These helpers are broadly divided into organic coenzymes, frequently derived from dietary vitamins, and robust inorganic cofactors.
Organic coenzymes heavily influence the efficiency of Enzyme Catalysis. They are further classified based on their binding affinity. Cosubstrates act as transient carriers, entering and leaving the active site just like standard substrates; familiar examples include NAD+ and ATP. In sharp contrast, prosthetic groups are tightly bound to the protein scaffolding, often covalently, acting as permanent fixtures in the active site. Heme groups are classic prosthetic groups, providing critical structural and chemical support for vital oxygen transport and complex electron shuttling.
The primary function of these organic helpers in Enzyme Catalysis is to act as group-transfer reagents. Molecules like Flavin Adenine Dinucleotide (FAD) utilize specialized ring structures, such as the isoalloxazine ring, to safely mediate dangerous one- or two-electron transfers, which are absolutely vital for oxidative phosphorylation. Meanwhile, inorganic cofactors such as the zinc ion in Carbonic Anhydrase deliberately activate water molecules for carbon dioxide hydration. These metal clusters systematically stabilize negative charges and induce structural conformational strain on substrates.
For medical students, this highly detailed slide highlights the strict biochemical rationale behind necessary vitamin supplementation. Vitamins are not merely abstract health boosters; they are the direct chemical precursors to the coenzymes explicitly required for cellular respiration and metabolism. A lack of Vitamin B, for instance, halts energy production because the biological catalysts lack the organic helpers they require to function properly.
Slide 11: Reversible Blockades in Enzyme Catalysis
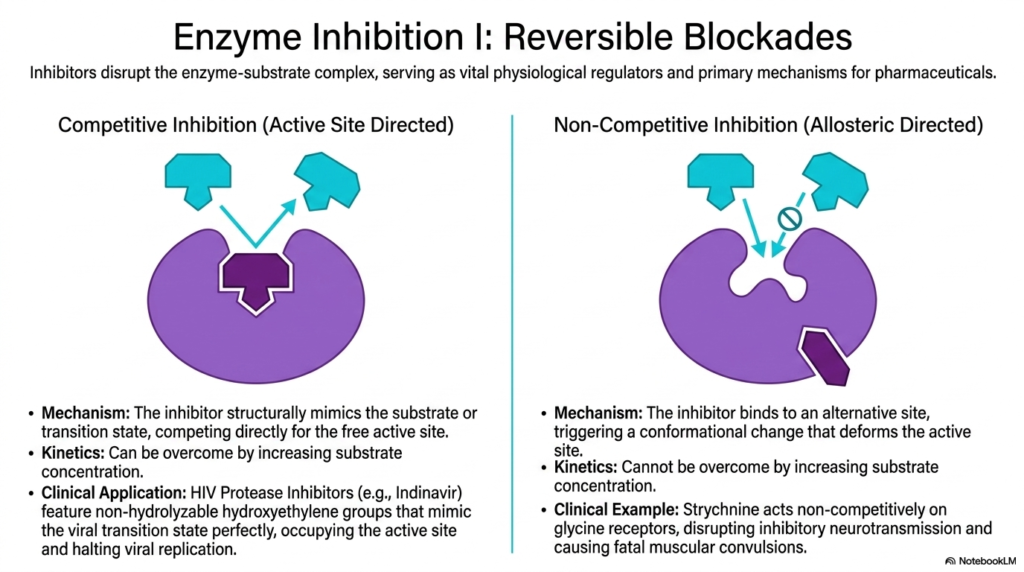
The strict regulation of biological activity is just as important as the acceleration of reactions. This slide introduces the vital physiological regulators and pharmacological cornerstones of Enzyme Catalysis: reversible inhibitors. These specialized molecules purposefully disrupt the enzyme-substrate complex through various mechanisms, providing vital dial-like control over cellular pathways. Competitive inhibition is uniquely active-site directed. The inhibitor structurally mimics the natural substrate or the transition state, aggressively competing for access to the free active site. This blockade can be overcome by increasing the substrate concentration to very high levels.
Competitive inhibitors represent a massive triumph in the pharmacological manipulation of Enzyme Catalysis. A prime clinical application discussed in the slide involves HIV Protease Inhibitors, such as the drug Indinavir. These brilliant medications feature specific non-hydrolyzable hydroxyethylene groups that perfectly mimic the viral transition state. By firmly occupying the active site with this rigid chemical mimic, the drug completely prevents the biological catalyst from processing viral proteins, effectively halting viral replication and safely managing the disease.
The slide sharply contrasts this with non-competitive, or allosteric-directed, regulation in Enzyme Catalysis. Here, the inhibitor binds to a completely distinct alternative site on the protein. This binding triggers a dramatic conformational change that physically deforms the active site from afar. Because the inhibitor and substrate do not compete for the same binding pocket, adding large amounts of substrate will not overcome a non-competitive blockade. The catalytic machinery is simply turned off regardless of local substrate availability.
A terrifying clinical example of this action is Strychnine poisoning. This deadly compound acts non-competitively on critical glycine receptors. By binding allosterically, it completely disrupts normal inhibitory neurotransmission, leading to unchecked neurological firing and fatal muscular convulsions. Understanding these contrasting mechanisms of inhibition is profoundly fundamental for modern toxicology and advanced drug design, as it dictates therapeutic dosing and overdose reversal strategies.
Slide 12: Irreversible Binding and Allostery in Enzyme Catalysis
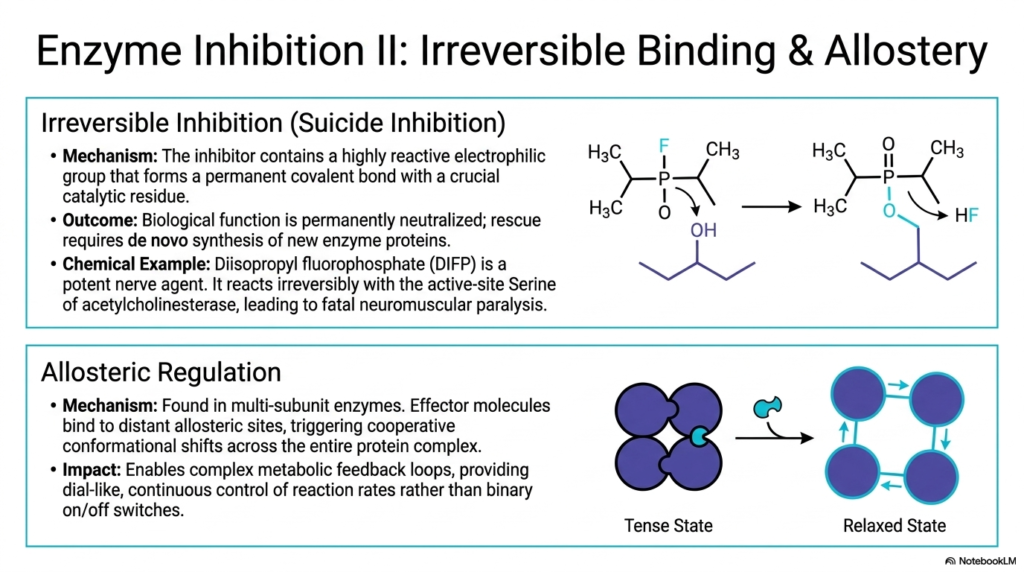
Expanding upon essential regulatory mechanisms, this slide explores irreversible blockades and cooperative binding dynamics. Irreversible inhibition, often termed suicide inhibition, completely neutralizes Enzyme Catalysis. The dangerous inhibitor utilizes a highly reactive electrophilic group to form a permanent, unbreakable covalent bond with a crucial catalytic residue within the active site. Once this powerful bond forms, the protein’s biological function is permanently destroyed. The cell cannot overcome this blockade; the only possible rescue is the slow, de novo synthesis of entirely new protein molecules.
The clinical and toxicological implications of shutting down Enzyme Catalysis so violently are immensely profound. The slide highlights Diisopropyl fluorophosphate (DIFP), a potent and terrifying nerve agent. DIFP reacts irreversibly with the specific active-site Serine residue of acetylcholinesterase. By permanently disabling the catalyst responsible for breaking down neurotransmitters, DIFP causes continuous, unstoppable muscle stimulation, ultimately leading to fatal neuromuscular paralysis. This chemical weapon perfectly illustrates the devastating, rapid consequences of permanently terminating vital biochemical pathways.
In sharp contrast to toxic irreversible binding, the slide also details the absolute elegance of normal allosteric regulation in Enzyme Catalysis. Found primarily in large, multi-subunit proteins, allostery utilizes unique effector molecules that bind to distant regulatory sites. This specific binding triggers complex, cooperative conformational shifts throughout the multi-protein complex, smoothly transitioning the structure between a rigid, inactive “Tense” state and a flexible, highly receptive “Relaxed” state. This unique cooperativity means that binding at one site heavily influences neighboring subunits.
This elegant structural shifting enables highly responsive, complex metabolic feedback loops. Instead of relying on simple binary on/off switches, natural allosteric regulation provides cellular systems with a highly sensitive, dial-like, continuous control over exact reaction rates. By dynamically responding to the changing concentrations of downstream products, these specialized biological catalysts meticulously balance energy production and biosynthesis in real-time.
Slide 13: Synthesizing Mechanics in Enzyme Catalysis
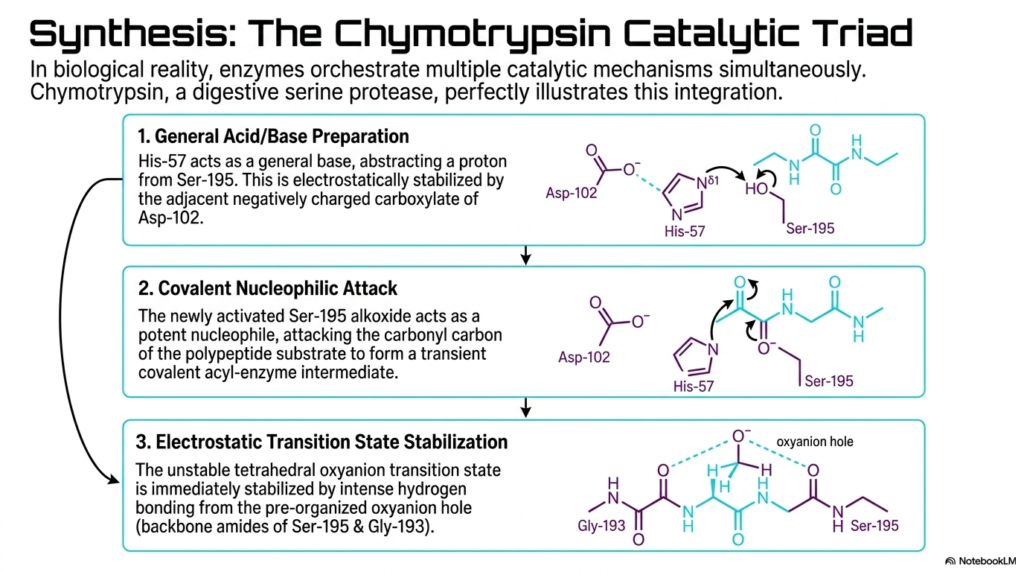
In true biological reality, these various mechanisms do not exist in isolation. This final synthesizing slide perfectly illustrates how proteins cleverly orchestrate multiple catalytic mechanisms simultaneously to achieve maximum Enzyme Catalysis. The chosen example is the remarkable Chymotrypsin catalytic triad, a sophisticated network found in digestive serine proteases. This highly conserved triad masterfully integrates general acid/base activation, covalent nucleophilic attack, and electrostatic transition-state stabilization into a seamless, elegant kinetic sequence. The sheer precision of this molecular machine relies on the perfect spatial alignment of three amino acids.
The first step of this integrated Enzyme Catalysis involves targeted general acid/base preparation. Histidine-57 acts as a powerful general base, abstracting a critical proton from its neighbor, Serine-195. This action is carefully electrostatically stabilized by the adjacent, negatively charged carboxylate group of Aspartate-102. By pulling the proton away, the Histidine rapidly activates the Serine, functionally converting its normal hydroxyl group into an extremely potent, highly reactive alkoxide nucleophile, perfectly primed for the very next step of the chemical reaction.
Following this rapid preparation, covalent nucleophilic Enzyme Catalysis immediately occurs. The newly activated Serine-195 attacks the carbonyl carbon of the incoming polypeptide substrate. This aggressive chemical strike forms a transient, covalent acyl-enzyme intermediate. Simultaneously, the intensely unstable tetrahedral oxyanion transition state is generated. This explosive intermediate is instantly pacified by intense hydrogen bonding from the pre-organized oxyanion hole, using the backbone amides of Serine-195 and Glycine-193 to provide substantial electrostatic transition-state stabilization.
By breaking down the entire Chymotrypsin mechanism step by step, college and medical students can finally see the grand synthesis of biochemical theory. The strict spatial alignment, the rapid shifting of protons, the deliberate formation of temporary bonds, and the exact stabilization of charges all happen within microseconds. This remarkably coordinated molecular dance represents the pinnacle of biological evolution, enabling complex organisms to digest food, replicate their DNA, and sustain life.
Please read our Content Disclaimer Statement.
Check out our social media channels: